|
Реферат на тему:
Равновесия в неводных растворах
Выполнила студентка 5го курса
Кекало Екатерина
Минск
Содержание:
Общая схема равновесий в растворах ………………………………………3
1. Ассоциативно-диссоциативные процессы………………………..….3
2. Образование продуктов присоединения …………………….……4
3. Ионизация………………………………………………………………7
4. Электролитическая диссоциация………………………………….….8
Влияние растворителя на равновесие в химических системах…………….9
1. Влияние растворителя на молекулярные ассоциативно-диссоциативные процессы…………………………………………………………………...9
2. Влияние растворителя на константы устойчивости комплексных соединений………………………………………………………………...11
Список литературы ………………………………………………………..…13
ОБЩАЯ СХЕМА РАВНОВЕСИЙ В РАСТВОРАХ
Процессы, сопряженные с образованием раствора, особенно электролитного, неизменно находились в центре внимания общей теории растворов. Именно вокруг вопросов, связанных с числом и характером стадий, предшествующих возникновению электролитного раствора, велись дискуссии между сторонниками различных теорий вопроса. Синтез этих двух основных направлений в учении о растворах в значительной мере был связан именно с установлением общей схемы равновесий в растворах.
1. Ассоциативно-диссоциативные процессы
Ассоциированная молекула растворенного вещества Аx
при растворении в растворителе S может претерпевать молекулярную
диссоциацию:
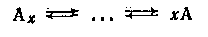
Последняя может быть связана с физическим влиянием растворителя (например, ослаблением межмолекулярного диполь-дипольного взаимодействия в растворителях с высокой диэлектрической проницаемостью). Однако большей частью энергия, нужная для разрыва связей в ассоциате, черпается из химического взаимодействия между Аx
и S:
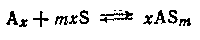
Если молекулы растворителя ассоциированы, то образование раствора приводит к изменению этого состояния тем больше, чем больше концентрация раствора. Молекулярная диссоциация растворителя может быть вызвана химическим взаимодействием между А и S:
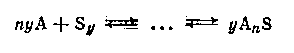
Определение степени ассоциации растворенного вещества в растворе относится к числу наиболее распространенных в практике физикохимического исследования экспериментов. В случае разбавленных растворов для решения этой задачи успешно применяют группу методов, основанных на законе Рауля - эбулиоскопию и, особенно, криоскопию. Большинство сведений о молекулярном состоянии растворенных веществ в разбавленных растворах, которыми располагает в настоящее время физическая химия, получены с помощью последнего метода. К недостаткам криоскопии и эбулиоскопии следует отнести сильное влияние на точность определения флуктуации концентрации в растворе (обстоятельство, которое не всегда учитывается исследователями), а также приложимость этих методов лишь к весьма разбавленным растворам.
Весьма полная информация о молекулярном состоянии растворенного вещества, особенно в тех распространенных случаях, когда образование ассоциатов обусловлено Н-связыо, может быть получено с помощью ИК (КРС)-спектроскопии. Уменьшение степени ассоциации, сопряженное с разрывом Н-связей, приводит к сдвигу частот валентных колебаний ОН в длинноволновую область. Во многих случаях установлена весьма определенная корреляция между сдвигом максимума полосы поглощения и степенью ассоциации. Впрочем, так же, как я в случае методов, основанных на законе Рауля, сколь-нибудь исчерпывающая количественная информация о степени ассоциации по спектроскопическим данным может быть получена в разбавленных растворах.
Ассоциативно-диссоциативные процессы в растворах с несколько более высокой концентрацией могут быть исследованы с помощью ЯМР-спектроскопии: разрыв ассоциатов через Н-связь приводит к сдвигу сигнала протонного магнитного резонанса в Сторону более сильного поля. (В системах, где компоненты содержат арильное ядро, это явление не проявляется с необходимой для точного расчета степени ассоциации четкостью).
С повышением концентрации раствора точность методов определения степени ассоциации быстро падает. Связано это не только с несовершенством экспериментальных и расчетных методик, которые основаны на недостаточном теоретическом обосновании методов, но прежде всего на возрастании по мере роста концентрации раствора степени неопределенности понятия «ассоциация». Действительно, в предельном случае — в индивидуальной жидкости— далеко не всегда бывает очевидным, какой критерий следует положить в основу понятия «степень ассоциации». Определенность, с которой физика вводит это понятие через представления о ближнем и дальнем порядках в жидкости, лимитируется отсутствием четкой границы между «порядками».
Для определения степени ассоциации индивидуальной жидкости предложено несколько методов. Весьма часто для этой цели применяют уравнение Рамзая — Шильдса, использующее данные по поверхностному натяжению у:
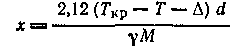
где х
—степень ассоциации; Ткр
— критическая температура; Т
— температура жидкости; Δ — константа, равная приблизительно 279°К; d
— плотность; М
— формульная молекулярная масса.
Близко по характеру входящих в него величин к данному уравнению примыкает уравнение Беннета — Митчелла
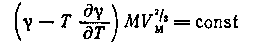
где VM
— мольный объем; а М
— истинная молекулярная масса.
2. Образование продуктов присоединения
(сольватация)
Переходя к рассмотрению следующей стадии равновесий в растворах, связанной со взаимодействием между компонентами раствора, необходимо, хотя бы кратко, остановиться на определении понятия «химическое взаимодействие в растворе».
Ответ на вопрос, что следует считать необходимым и достаточным признаком для суждения об отсутствии химического взаимодействия между компонентами раствора с самого начала был связан с трудностями методологического характера. Как известно, не может быть дано сколь-нибудь строгого определения негативного понятия, а отсутствие
взаимодействия несомненно принадлежит к числу таковых.
Далее, если считать признаком возникновения химической связи взаимодействие электронных облаков атомов химических элементов, то строго говоря, поскольку плотность электронного облака на любом конечном расстоянии от атомного ядра не равна нулю, следует признать наличие химического взаимодействия между любыми двумя атомами, находящимися друг от друга на любом конечном расстоянии. Но, очевидно также и то, что исповедование этого положения лишило бы химию сколь-нибудь четких критериев в определении самого ее фундаментального понятия. Поэтому в определении понятия химическое взаимодействие (так же, как и атомный радиус) всегда лежит какое-либо предварительно оговоренное условие, а само оно всецело зависит от этого условия.
По-видимому, наиболее удобным критерием, который мог бы служить для суждения о наличии или отсутствии химического взаимодействия в химической системе, является энергия взаимодействия. Можно было бы выбрать такую минимальную ее величину, которая могла бы быть отнесена к химическому процессу. Однако несовершенство и значительная сложность методов определения энергии взаимодействуя не позволяет в настоящее время считать такой выбор удачным.
Тип связи также не может служить критерием, который лег бы в основу суждения о наличии или отсутствии взаимодействия. В самом деле, между крайними случаями — образованием чисто ковалентной или ионной связи и слабым вандерваальсовским взаимодействием — существует множество промежуточных типов, одна классификация которых подчас вызывает значительные затруднения.
Строго говоря, процесс молекулярной ассоциации (диссоциации) должен быть отнесен к химическому взаимодействию. Действительно, если, например, ассоциат (АН)Х
через Н-связь подвергается молекулярной диссоциации, то это сопряжено с разрывом Н-связи, т. е. по всем формальным признакам (в число которых включается и весьма значительная величина энергии связи) подходит под химическое взаимодействие. Однако ассоциативно-диссоциативные процессы не принято относить к химическому взаимодействию. В противном случае пришлось бы разграничивать химическое взаимодействие, относящееся к первой стадии равновесий в растворах и межмолекулярное взаимодействие между компонентами раствора:
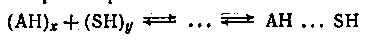
(предполагается для простоты, что молекулы растворителя также ассоциированы через Н-связь).
.Впрочем, вряд ли было бы целесообразным считать любое межмолекулярное взаимодействие данного типа химическим. Так, если АН и SH — соединения тождественной химической природы, стоящие к тому же близко в гомологическом ряду (например, пропанол и бутанол, изомеры крезола, валериановая и масляная кислоты и т. п.), то образование новых межмолекулярных связей (перекрестных ассоциатов) не может быть отнесено к химическому взаимодействию, хотя бы потому, что это взаимодействие практически не сопряжено с таким изменением энергетических характеристик системы, которое могло бы быть с уверенностью зафиксировано.
Выбор критерия, которым следует пользоваться для суждения о взаимодействии в химической системе, в каждый данный период развития химии зависит от развития теория, а также методов исследования химических систем. Так, в настоящее время благодаря интенсивному развитию ИК- и радиоспектроскопии, появляется возможность получения гораздо более интимных сведений о химической системе, чем два-три десятилетия назад. И несомненно, что спустя определенный промежуток времени появятся методы исследования, которые углубят степень познания процессов, протекающих в системе, столь же значительно, как это сделали упомянутые методы. Вот почему всегда, говоря о наличии или отсутствии химического взаимодействия, следует оговаривать, о какой степени приближения идет речь, так как очевидно, что универсального определения, одинаково пригодного для всех случаев, быть не может.
Хотя современные методы исследования позволяют зафиксировать весьма слабые химические взаимодействия с энергией порядка десятой доли ккал/моль,
в подавляющем большинстве случаев в растворах они не могут быть с уверенностью обнаружены. Дело в том, что энергетические эффекты смешения соизмеримы, а зачастую значительно превышают указанную величину. Вот почему слабая энергетика химического взаимодействия в растворах не может быть выделена на фоне значительных тепловых эффектов смешения, а также теплового движения молекул.
В этих случаях предпочитают говорить'об отсутствии химического взаимодействия между компонентами, образующими раствор. Такая точка зрения является приближением, весьма пригодным для рассмотрения процессов, протекающих в растворах.
Таким образом, процесс химического взаимодействия между растворенным веществом АК и растворителем S

с точки зрения -представлений, которые будут соблюдаться и в этой книге, протекает не всегда. Он необходим лишь тогда, когда образуется электролитный раствор. В литературе химическое соединение (АК)m
Sn
часто называют сольватом, а сам процесс — сольватацией. Прежде понятие «сольват» очень часто отождествляли с понятием «соединение неопределенного состава», В тех же случаях, когда стехиометрия взаимодействия между АК и S была точно известна, употребляли термин «продукт присоединения». По мере усовершенствования методов исследования растворов смысловое различие между этими двумя понятиями постепенно исчезает. В этой книге мы будем считать эти понятия синонимами, применяя, следуя традиции, укоренившейся в литературе, термин «сольватация» для обозначения процессов присоединения молекул растворителя к ионам, а термин «продукт присоединения» — для сольватов, образующихся в жидких системах. В случае ионных сольватов принято различать первичную и вторичную сольватные оболочки. При этом в первичную оболочку входит то сравнительно небольшое количество молекул растворителя, которые непосредственно связаны с ионом (причем энергия связи ион — молекула растворителя соизмерима с энергией химической связи) и которые перемещаются в растворе вместе с ионом. Вторичная сольватная оболочка включает значительно большее число молекул растворителя. Границы этой оболочки определяются изменением физико-химических характеристик растворителя под влиянием иона.
Исследование стехиометрии и глубины взаимодействия в растворах, т. е. изучение стехиометрии и константы равновесия процесса является предметом обширнейшего раздела теории растворов.
3. Ионизация
Энергетический анализ процесса электролитической диссоциации показывает, что сольват (AK)m
Sn
не может непосредственно перейти в диссоциированные на ионы соединения. Так, продукт присоединения амина к кислоте RCOOH • NR3
’
не может непосредственно дать ионы RCOO-
и [NR3
H]+
. Для этого потребовалось бы затратить весьма значительную энергию, необходимую для разрыва связи —О-Н. Поэтому стадии электролитической диссоциации продукта присоединения амина к кислоте предшествует, стадия ионизации продукта присоединения, заключающаяся в отторжении протона от гидроксильной группы с переходом его на атом азота, причем образуется ионизированный комплекс RCOO-
-[NR3
H]+
.
В общем виде процесс ионизации передается схемой:
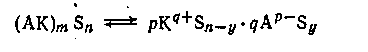
Нередко взаимодействие между компонентами раствора может ограничиваться стадией ионизации. Особенно часто это встречается в растворах с низкой диэлектрической проницаемостью. Например, при сливании разбавленных бензоловых растворов трихлоруксусной кислоты и триэтиламина образуется практически нацело ионизованный продукт присоединения, однако электропроводность раствора очень низка.
Наиболее удобным методом изучения стадии ионизации в растворах является исследование колебательных спектров. Однако константа ионизации при этом может быть рассчитана лишь тогда, когда электролитическая диссоциация протекает в малой степени, так как в подавляющем большинстве случаев на ИК-спектрах полосы поглощения, отвечающие ионам, находящимся и в свободном состоянии и в ионизованном комплексе, неразличимы.
В жидких системах информация о стадии ионизации может быть получена на основании кондуктометрических измерений.
В растворителях с высокими диэлектрическими проницаемостями электролитическая диссоциация может протекать полностью и равновесная концентрация ионизованного комплекса будет пренебрежимо мала. Однако это не означает, что электролитный раствор мог образоваться, минуя стадию ионизации. Развитие методов изучения быстрых реакций показывает, что даже в растворителях с весьма высокими диэлектрическими проницаемостями (вода, серная кислота) возникновению свободных ионов неизбежно предшествует стадия ионизации.
4. Электролитическая диссоциация
.
Ионизация сольвата является необходимым, но еще не обязательным условием образования электролитного раствора. Обязательным же условием диссоциации ионизированного комплекса на ионы является достаточно высокая диэлектрическая проницаемость раствора. Лишь в этом случае ионизованный комплекс распадается на ионы:

Несмотря на то, что полная ионизация протекает достаточно часто, полная электролитическая диссоциация (сильные электролиты) наблюдается значительно реже. Абсолютно подавляющее большинство изученных электролитных неводных растворов образовано слабыми электролитами, реже — электролитами средней силы.
Основные методы определения константы электролитической диссоциации обычно сводятся к кондукто- либо потенциометрическим методикам и приложимы лишь к весьма разбавленным растворам. Другие методы определения констант диссоциации (например, спектрофотометрические) имеют ограниченное применение.
Современный арсенал методов исследования растворов позволяет почти всегда уверенно определять число стадий для каждого конкретного объекта.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ НА РАВНОВЕСИЕ В ХИМИЧЕСКИХ СИСТЕМАХ
Проблема влияния растворителя на равновесие процессов, протекающих в химических системах, сводится в первую очередь к рассмотрению двух вопросов — как изменяется равновесие процесса при переходе из газовой фазы в данный растворитель и от; одного растворителя к другому.
Влияние физических свойств растворителя на константы равновесия процессов в химических системах может быть объяснено с электростатических позиций, если учесть, что в значительном числе случаев в первом приближении химические процессы сводятся к электростатическому взаимодействию, константа равновесия которого описывается уравнением Борна:
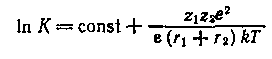
Из уравнения следует, что из физических свойств растворителя, определяющих константу равновесия, основным является диэлектрическая проницаемость. Действительно, почти во всех случаях можно установить весьма простую корреляцию между константами равновесий разнообразных процессов и диэлектрической проницаемостью.
Влияние диэлектрической проницаемости на равновесие в химических системах может быть установлено и в случае взаимодействия двух диполей.
1. Влияние растворителя
на молекулярные ассоциативно-диссоциативные процессы
Во многих случаях молекулярная диссоциация ассоциированных компонентов, образующих жидкий раствор, обусловлена химическим взаимодействием между ними.

Табл.1. Константы равновесия и теплоты процесса (в кДж/моль) диссоциации димеров уксусной кислоты в различных растворителях
В табл.1 приведены значения констант равновесия про цесса распада димеров уксусной кислоты B паре и различных растворителях. Несмотря на то, что таблица составлена по данным различных авторов и данные эти относятся к довольно широкому интервалу температур, можно установить четкую корреляцию между ассоциированным состоянием уксусной кислоты и химическими свойствами растворителя. В паре молекулы уксусной кислоты практически полностью являются димерными. Во всех растворителях уксусная кислота находится в виде равновесной смеси димерных и мономерных молекул. Нетрудно заметить, что на положение равновесия диэлектрическая проницаемость оказывает гораздо меньшее влияние, чем химические свойства растворителей. Так, в растворителях, характеризующихся весьма близкими значениями ε — гексане,
сероуглероде, четыреххлористом углероде, бензоле и диоксане, величины Кмон
различается весьма существенно. В химически инертных по отношению к уксусной кислоте растворителях равновесие практически смещено в сторону димера. Величина Кмон
в бензоле приблизительно на порядок выше, чем в остальных инертных растворителях, что обусловлено взаимодействием (впрочем, довольно слабым) кислоты с бензолом по π-связям последнего. В диоксане же, который является растворителем с отчетливо выраженными основными свойствами, равновесие уже значительно смещено в сторону мономера.
Химические свойства растворенного вещества также в значительной степени определяют его ассоциативное состояние. Так, величины Кмон
растворов уксусной, монохлоруксусной и трихлоруксусной кислот в диоксане, рассчитанные по криоскопическим данным составляют 0,8; 24 и ∞, т. е. увеличиваются по мере увеличения степени взаимодействия кислот с растворителем.
Причиной, обусловливающей распад ассоциированных молекул многих веществ, ншшстся в большинстве случаев образование Н-связи между компонентами раствора. Как известно, мерой прочности Н-связи (а, соответственно, и степени распада ассоциатов) является сдвиг максимума полосы поглощения, относящейся к группировке, которая участвует в образовании Н-связи. Спектроскопическая литература изобилует примерами, иллюстрирующими эту закономерность.
Влияние растворителя на ассоциативное состояние растворенного вещества связано прежде всего с ε. Это влияние отчетливо сказывается лишь в тех растворителях, энергией взаимодействия которых с растворенным веществом можно пренебречь по сравнению с энергией связи молекул в ассоциате. Можно установить влияние растворителя и на константу равновесия конформационных превращений молекул в растворах. Поскольку последние сопряжены с изменением дипольного момента, молекулы, следует ожидать влияния ε на константу равновесия этого процесса.
В литературе имеются весьма скудные данные по влиянию растворителя на константу равновесия процессов образования продуктов присоединения  . Исследования в этом направлении охватывают ограниченный круг растворителей в небольшом интервале диэлектрических проницаемостей; кроме того, не всегда удается разграничить влияние физических и химических факторов. . Исследования в этом направлении охватывают ограниченный круг растворителей в небольшом интервале диэлектрических проницаемостей; кроме того, не всегда удается разграничить влияние физических и химических факторов.
2. Влияние растворителя
на константы устойчивости комплексных соединений
Рассмотрим вопросы, связанные с изменением константы равновесия (константы устойчивости) Кк
процессов комплексообразования, т. е. процессов присоединения к иону металла иона (в частном случае нейтральной молекулы) лиганда
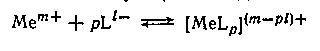
при переходе от одного растворителя к другому, либо с изменением состава смешанного растворителя.
Многочисленные литературные данные свидетельствуют, что при переходе от одного растворителя к другому состав комплексного иона может изменяться — вследствие внедрения молекул растворителя во внутреннюю сферу комплекса
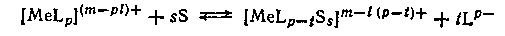
или замены внедрившихся молекул одного растворителя молекулами другого

Изменение констант устойчивости комплексных, соединений с изменением растворителя (так далее будет называться переход от одного растворителя к другому, либо изменение состава двойного смешанного растворителя) обусловлено изменением как физических, так и химических характеристик среды. Кроме того, в случае комплексных соединений добавляется еще один специфический фактор, играющий большую роль в определении степени, с которой растворитель влияет на изменение Кк
—
это характер связи между центральным атомом и лигандом.
Для объяснения влияния физических свойств растворителя и, прежде всего, диэлектрической проницаемости на прочность комплекса в первом, а в большинстве случаев достаточно хорошем, приближении можно ограничиться представлениями о том, что взаимодействие данного типа подчиняется закономерностям электростатического взаимодействия.
Влияние сольватационной способности растворителя на величины Кк
хорошо иллюстрируются примерами растворов роданидных комплексов металлов. Константы устойчивости этих комплексов в весьма близких по величинам ε
растворителях (метанол, ДМФ и ацетонитрил) различаются на много порядков. Кроме того, прочность комплексов в ацетонитриле значительно выше, чем в метаноле и ДМФ — в соответствии с гораздо меньшей сольватационной способностью первого по сравнению с последними.
Иногда изменение растворителя ведет к существенному изменению природы лиганда. Так, например, глицин, который в диоксане находится в молекулярной форме, в воде находится в форме 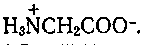 Поэтому константа устойчивости комплекса никеля с глицином в смешанном растворителе вода — диоксан изменяется чрезвычайно сильно, в то время как константы устойчивости комплексов никеля с лигандами, молекулярное состояние которых не зависит от растворителя, в этом смешанном растворителе изменяются гораздо меньше. Поэтому константа устойчивости комплекса никеля с глицином в смешанном растворителе вода — диоксан изменяется чрезвычайно сильно, в то время как константы устойчивости комплексов никеля с лигандами, молекулярное состояние которых не зависит от растворителя, в этом смешанном растворителе изменяются гораздо меньше.
Тесная зависимость характера изменения константы устойчивости комплекса от растворителя вытекает уже из следующей элементарной предпосылки: чем выше степень ионности связи, тем влияние ε должно сказываться сильнее. В общем случае изменение устойчивости комплексов со связью Ме…..О с изменением ε более резкое, чем в комплексах со связью Me……N. Иногда уменьшение ε приводит к уменьшению устойчивости комплексов со связью Me…..N — явление, которое не свойственно в общем случае комплексам со связью Me…..О.
Так же растворитель влияет на таутомерные равновесия, константы электролитической диссоциации, силу электролитов, кислотность неводных растворов и т.д.
Список литературы:
1. Мелвин-Хьюз Е. Равновесие и кинетика реакций в растворах. Л.Химия. 1975.
2. Мейтис Л. Введение в курс химического равновесия и кинетики. М.Мир.1984.
3. Баттлер Д, Ионные равновесия. Л. Химия. 1973,
4. Булатов М.И. Расчеты равновесий в аналитической химии. М. Химия. 1984.
5. Хартли Ф., Бергес К., Олкок Р. Равновесие в растворах, М.Мир, 1983.
6. Фиштик И.Ф. Термодинамика сложных химических равновесий. Кишинев. Штиинца. 1989.
7. Комарь Н.П. Гомогенные ионные равновесия. Харьков. Вита школа. 1983.
8. Россотти Ф,, Россотти X. Определение констант устойчивости и других констант равновесия в растворах. М. Мир. 1965.
9. Кугаевский А.А. Расчет равновесий в растворе. Харьков. Высшая школа. 1980.
10. Ю.Я, Фиал ко в, А,Н.Житомирский, Ю.А.Тарасенко. Физическая химия неводных растворов.
|