| Введение
В настоящее время в органической химии синтезировано более 20 миллионов химических веществ, набор сведений о которых весьма обширен. Активное использование в химических исследованиях этой огромной информации невозможно без привлечения средств вычислительной техники. Компьютерные технологии и математическое моделирование позволяют отказаться от традиционного метода поиска химических веществ с заданными свойствами путем экспериментов, которые являются чрезвычайно сложными, длительными и дорогостоящими. Так, разработка единственного лекарственного препарата требует в среднем от 8 до 20 лет, а затраты составляют до 100 миллионов долларов. Согласно статистике, удачным оказывается приблизительно одно из 10 тысяч испытаний возможных лекарственных препаратов. Исключительно большое число структур, которые необходимо исследовать, может быть проиллюстрировано тем фактом, что одна структура и 20 заместителей, присоединенных в шести различных положениях, будут приводить к полному числу исследуемых структур, равному 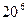 . В связи с этим многие фирмы связывают успех в разработке новых препаратов с внедрением в химические исследования компьютерного и математического моделирования, резко сокращающего сроки создания физиологически активных веществ, путем отсеивания заведомо неактивных и высокотоксичных соединений. Поэтому неудивительно, что в настоящее время для достижения такой заманчивой цели прилагаются значительные усилия. Например, расходы фирмы «Pfizer» на научные исследования по поиску фармацевтических препаратов в 1990 году возросли более чем на 20% и составили 640 миллионов долларов. По оценке «International Resource Development Inc» особенно быстро растет объем финансирования работ по созданию компьютерных систем для моделирования различных свойств химических веществ – от 66 миллионов долларов в 1983 голу до 8,5 миллиардов долларов в 1993 году. При создании таких систем возникают сложные, выходящие за рамки информационного поиска задачи, решение которых требует разработки специальных методов и моделей, оригинальных алгоритмов и соответствующего программного обеспечения. При этом подобные системы будут обладать рядом преимуществ, обусловленных мощной информационной поддержкой в виде фактографических баз данных, наличие которых позволит наряду с задачами информационного характера, решать и такие важные в научном и практическом отношении проблемы, как установление эмпирических закономерностей для моделирования связи между строением вещества и его физико-химическими и биологическими свойствами. Такого рода системы позволят более объективно использовать накопленный экспериментальный материал в виде разнообразных зависимостей «структура соединения – физико-химические и биологические свойства». . В связи с этим многие фирмы связывают успех в разработке новых препаратов с внедрением в химические исследования компьютерного и математического моделирования, резко сокращающего сроки создания физиологически активных веществ, путем отсеивания заведомо неактивных и высокотоксичных соединений. Поэтому неудивительно, что в настоящее время для достижения такой заманчивой цели прилагаются значительные усилия. Например, расходы фирмы «Pfizer» на научные исследования по поиску фармацевтических препаратов в 1990 году возросли более чем на 20% и составили 640 миллионов долларов. По оценке «International Resource Development Inc» особенно быстро растет объем финансирования работ по созданию компьютерных систем для моделирования различных свойств химических веществ – от 66 миллионов долларов в 1983 голу до 8,5 миллиардов долларов в 1993 году. При создании таких систем возникают сложные, выходящие за рамки информационного поиска задачи, решение которых требует разработки специальных методов и моделей, оригинальных алгоритмов и соответствующего программного обеспечения. При этом подобные системы будут обладать рядом преимуществ, обусловленных мощной информационной поддержкой в виде фактографических баз данных, наличие которых позволит наряду с задачами информационного характера, решать и такие важные в научном и практическом отношении проблемы, как установление эмпирических закономерностей для моделирования связи между строением вещества и его физико-химическими и биологическими свойствами. Такого рода системы позволят более объективно использовать накопленный экспериментальный материал в виде разнообразных зависимостей «структура соединения – физико-химические и биологические свойства».
Таким образом, проблемы создания информационно-математической инфраструктуры системы научных химических исследований, в первую очередь комплекса проблемно-ориентированных баз данных, по полноте адекватных нуждам пользователей, является в настоящее время ключевой для интенсификации научных исследований. Изложенное выше определяет актуальность, научную и практическую значимость проведенных в монографии исследований в одной из наиболее важных областей химической информатики – разработке компьютерных средств и математических методов для моделирования связи между строением вещества и его свойствами. Вряд ли необходима более развернутая аргументация в пользу актуальности и важности компьютерно – моделирующих систем с обучением для таких предметных областей, как химия, биология, медицина и другие науки о жизни, где постановка реального или лабораторного эксперимента либо изрядно затруднена, либо вообще невозможна по тем или иным соображением. Хотелось бы отметить, что поиск новых высокоактивных и безопасных для человека и окружающей среды химических препаратов с заранее заданными свойствами является важнейшей фундаментальной проблемой мировой науки, так как создание таких веществ есть одно из основных условий роста технологической мощи современного общества.
Цель данной работы состоит в разработке эффективных методов математического и компьютерного моделирования связи «структура химических соединений –молекулярные свойства и биологическая активность» на основе стратегии формирования базы данных и знаний из имеющихся примеров, а также в создании компьютерных технологий поддержки профессиональных химико-структурно-биологических баз данных и знаний. Для достижения указанной цели по поиску эффективных препаратов с заданными свойствами требуется углубленный анализ и теоретические исследования первичных экспериментальных данных с использованием современных информационно - компьютерных технологий и методов математического моделирования.
Постановка задачи
Современное развитие общества связано с перевооружением всех отраслей народного хозяйства на основе современных достижений науки и техники. Особенно важную роль в решении этой задачи будет иметь внедрение новой информационной технологии, реализуемой в прикладных информационных системах на базе средств вычислительной техники. Автоматизированные информационные системы (информационно-поисковые, управляющие, диагностические, моделирующие) предназначены для оперирования особо интенсивными потоками информации, отсутствие или недостаток которой ведет к неэффективным решениям, сопровождаемым различными негативными последствиями. Их техническая и программная части должны обеспечивать выполнение специальных процедур автоматизированного сбора, хранения и обработки. В области науки большое значение имеет также создание математического обеспечения (методов, моделей, алгоритмов и программ) для моделирования, анализа и прогнозирования зависимостей типа структура объектов – его свойства, в частности, молекулярная структура – физико-химические свойства и биологическая активность. Таким образом, можно говорить о необходимости создания информационно – математической инфраструктуры в конкретных областях научных исследований. С этой глобальной задачей тесно связаны более локальные задачи создания автоматизированных систем научных исследований (АСНИ) по поиску препаратов с заданными свойствами в области органической химии, в которой огромный объем информации, принципиальная сложность и новизна решаемых научных проблем как раз и выдвигают в качестве одной из первоочередных задач разработку высокоэффективных средств информационно – компьютерной поддержки. Следует также подчеркнуть, что помимо прикладной роли, разработка таких систем имеет большое самостоятельное значение. Более того, можно утверждать, что без разработки эффективных АСНИ в органической химии, успешное решение проблемы поиска препаратов с заданными свойствами будет вообще невозможно, потому что возникают различные проблемы, в том числе, проблема оценки адекватности результатов моделирования при изучении явлений, экспериментальный анализ которых обычными средствами, как правило, невозможен. Так, при исследованиях по химическому канцерогенезу обычными лабораторными средствами, характерное время эксперимента – около трех лет, а стоимость изучения одного соединения – около 12 миллионов долларов США. Если учесть, что к настоящему времени известны более 15 миллионов химических веществ, число которых ежегодно увеличивается на 500 тысяч вновь созданных, то утверждение о затрудненности экспериментального анализа всего потока обычным путем становится просто очевидным [5]. Аналогично, эффективность работ по поиску новых химических средств защиты растений (ХСЗР) обычными средствами (путем синтеза и массовых испытаний большого числа самых различных химических соединений) чрезвычайно мала, так как для создания конкурентноспособного нового действующего вещества необходим синтез и биологические испытания от 30 до 100 тысяч химических соединений [6], причем, стоимость разработки нового продукта без затрат на создание промышленного производства составляет 10 – 35 миллионов долларов, а время, затрачиваемое на разработку, составляет около 10 лет. Поэтому, применение математических методов и АСНИ для установления количественных соотношений структура – активность и их использование для прогнозирования потенциально активных соединений становятся одними из важных и основных путей изыскания препаратов с заданными свойствами, так как они приведут к существенному сокращению времени и объема поисковых работ, а следовательно, и затрат на разработку.
Основными функциями АСНИ являются: сбор, автоматизированная обработка экспериментальных данных или другой информации, получение и исследование математических моделей, изучаемых объектов, явлений, процессов, с целью использования их в дальнейшем для получения новых знаний. Характерной тенденцией развития автоматизированных информационных систем является соединение в них средств удовлетворения чисто информационных потребностей (поиск в базах данных) и средств переработки информации (получение нового знания), создание которых связано с программной реализацией существующих и разработкой новых математических моделей и алгоритмов обработки данных.
Ядром АСНИ является информационно – математическое обеспечение, включающее информационно – вычислительные системы и системы математического моделирования и обработки данных на основе средств вычислительной техники. В состав информационного обеспечения научных химиико - биологических исследований в глобальном аспекте должны входить [7-14]:
· библиография по проблеме поиска химических веществ с заданными свойствами;
· результаты биологических испытаний в системах скрининга химических соединений;
· химико-структурно-биологическая информация из патентных и литературных источников;
· данные по метаболизму, токсикологии и экологии органических соединений;
· экономика производства и применения, мировой ассортимент действующих веществ;
· межотраслевая информация (почвы, климатические условия, культуры, площади, вредители и т.д.);
· сырье и реактивы;
· физико – химические свойства органических веществ;
· спектральные и структурные данные химических соединений;
· химизм процессов, технологические параметры, схемы и материальные балансы;
· аппаратура.
Современный этап развития информационного обеспечения работ по поиску препаратов с заданными свойствами характеризуется созданием и эксплуатацией автоматизированных баз данных (БД) по некоторым из перечисленных аспектов, однако по большинству аспектов разработка БД ведется в стране недостаточно интенсивно. В первую очередь это связано с отсутствием должной координации работ, их недостаточным финансированием, значительной трудоемкостью и рутинным характером.
Математическое обеспечение разработок в химических и биологических исследованиях по поиску препаратов с заданными свойствами требует создания методов, моделей. Алгоритмов и программного обеспечения по следующим основным аспектам [4, 15 - 43]:
1. Ввод структурной информации в ЭВМ (рисование молекулярной структуры на дисплее и порождение ее машинного представления).
2. Унификация биологических данных (моделирование индексов активности типа 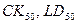 из исходных показателей биологической активности). из исходных показателей биологической активности).
3. Порождение описания молекулярной структуры, исходя из матрицы смежности.
4. Конформационные и квантовохимические расчеты (расчеты пространственного и электронного строения молекул).
5. Анализ и прогнозирование биологической активности (реализация различных методов, моделей и алгоритмов моделирования связи структура – активность).
6. Генерация химических веществ исходя из заданных базисных структур и/или фрагментов и ограничений на их модификацию.
7. Оптимизация скрининга (информационно – вычислительная поддержка и управление системой скрининга химических соединений на биологическую активность).
8. Планирование и обработка эксперимента методами многомерного статистического анализа и моделирования данных.
9. Расчет свойств веществ (прогнозирование физико – химических, в том числе пожаровзрывоопасных и токсикологических, характеристик индивидуальных органических соединений).
10. Расчет процессов, аппаратов, технологических схем (оптимизация способов получения продуктов и аппаратурного оформления процессов).
11. Технико – экономические исследования (прогнозная технико – экономическая оценка научно – исследовательских и опытно – технологических работ, промышленного производства и продуктов, оптимизация ассортимента).
12. Оформление и документация (средства подготовки выходных документов в удобном для пользователей виде).
Данная работа направлена на решение вышеперечисленных задач по пунктам 1, 2, 3, 5, 6, 8, 9, 12 в рамках автоматизированной системы научных исследований в органической химии. АСНИ подобного рода позволят радикально интенсифицировать научные исследования в этой области. Эффект от создания АСНИ возникает за счет [4, 10, 12, 15 – 16, 21 – 25, 27, 30, 38 - 43]:
· уменьшения трудозатрат на поиск нужной информации;
· сокращения объема пустых синтезов и биологических испытаний на основе использования математических методов анализа и прогнозирования биологически активных соединений (отказ от синтеза и биологических испытаний химических веществ, с высокой вероятностью являющихся неактивными или высокоопасными);
· использования системы компьютерной генерации потенциально – активных структур для планирования синтеза, исходя из прототипов и их возможных структурных модификаций;
· сокращения экспериментов, связанных с поисковым синтезом в рядах химических соединений;
· сокращения затрат на изучение токсикологии, экотоксикологии, экологии путем использования компьютерной системы моделирования токсических свойств органических веществ;
· сокращения объема экспериментальных исследований на основе компьютерной оценки физико–химических характеристик исследуемых соединений.
Основные этапы и ожидаемые результаты.
1. Разработка непараметрического подхода к моделированию зависимости «доза - эффект».
Проблема исследования зависимости проявления эффекта от дозы яда, лекарственного препарата, проникающей радиации или другого повреждающего фактора является основополагающей в токсикологии, фармакологии, радиобиологии, биохимии, микробиологии, эпидемиологии и в других областях медицины и биологии.
На современном этапе токсикометрии используются величины эффективных доз, вызывающих появление учитываемого эффекта в экспериментальной группе тест - объектов с заданной вероятностью: 0,05; 0,016; 0,5; 0,84; 0,95. Такие дозы получили название эффективных доз: ED5
, ED16
, ED50
, ED84
, ED95
. Общая зависимость вероятности появления эффекта от воздействия заданной дозы определяется как функция эффективности. В качестве единого показателя для сравнения тестируемых препаратов принята среднеэффективная доза (ED50
), определяемая из функции эффективности. Трудность заключается в нахождении статистически обоснованных расчетно-экспериментальных значений категорий эффективных доз, которые представляют собой случайные величины. Проблема вероятностной оценки токсического и других эффектов рассматривается как одна из важнейших в токсикометрии и других разделах биологии и широко разрабатывается исходя из задач экспериментальной практики.
Открытие Гауссом закона нормального распределения послужило основой для разработки пробит–анализа, который в разных модификациях используется до настоящего времени в качестве основного (традиционного) метода определения среднеэффективных доз. В токсикометрии особенно важное значение отводится методам определения среднеэффективных доз, так как они являются теми решающими факторами, от которых зависит способ планирования экспериментов, порядок формирования и объем исходных данных, а в конечном итоге качество, эффективность и достоверность искомых показателей токсичности. По этим признакам проблему токсикометрической оценки показателей токсичности можно рассматривать как фундаментальную проблему теоретической токсикологии, имеющей прикладное значение для других разделов биологии и медицины.
Необходимо отметить, что для корректного применения методов пробит – анализа необходимым условием является нормальность распределения функции эффективности. Однако, в традиционных методах определения среднеэффективных доз критерии оценки нормальности функции эффективности отсутствуют, что ставит под сомнение универсальность этих методов для любой экспериментальной ситуации.
Принимая во внимание приведенные доводы, становится ясно, что современная методология токсикологического эксперимента и токсикометрии требует разработки таких моделей и подходов в оценке показателей токсичности, которые бы не предъявляли каких-либо граничных условий к планированию и выполнению токсикологического эксперимента, позволяли бы использовать для нахождения конечных оценок результаты независимых единичных испытаний и основывались на законах математической статистики и теории вероятностей.
Разработка новых токсикометрических моделей оценки токсичности должна быть направлена на максимально возможное уменьшение числа токсикологических испытаний на живых тест – объектах при сохранении заданной надежности конечных показателей токсичности, то есть на оптимизацию токсикологического эксперимента и сокращения материальных затрат. Предлагается исследовать возможности непараметрических методов для оценивания функции эффективности и провести сравнение их с традиционными подходами.
Тем самым, в рамках вышеописанной задачи, необходима разработка непараметрического метода оценки функции эффективности и реализация компьютерной поддержки этого метода.
2. Разработка универсальной модели классификации химических соединений по показателю токсичности
Следствием разнообразия методологических подходов к решению проблемы классификации токсикантов является разнообразие самих классификаций в различных странах. Наиболее широкое применение находят классификации, направленные на обеспечение безопасности работающих и безопасности массового потребителя. К указанным классификациям относится классификация опасных веществ, применяемая в странах ЕЭС, классификация вредных веществ, согласованная в рамках восточноевропейских стран, классификации для целей гигиены труда, используемые в Китае, Мексике, Финляндии и других странах. Достаточно хорошее соответствие существует между критериями острой токсичности в классификациях ЕЭС, СЭВ и Китая, что продемонстрировано таблицах ниже [13].
Следует иметь в виду, что в странах ЕЭС не устанавливается класс для малотоксичных соединений и те вещества, для которых LD50
при введении в желудок и при нанесении на кожу превышает 2000 мг/кг и CL50
превышает 20000 мг/м3
, формально не попадают ни в один из утвержденных классов.
Таблица 1. Классификация химических соединений по токсичности, принятая в cтранах ЕЭС.
| Показатель
|
Классы токсичности веществ
|
| I
очень токсичные
|
II
токсичные
|
III
вредные
|
| LD50
(мг/кг)
Введение в желудок
|
<25
|
25-200
|
200-2000
|
| LD50
(мг/кг)
Нанесение на кожу
|
<50
|
50-400
|
400-2000
|
| CL50
(мг/м3
)
Ингаляционное воздействие
|
<500
|
500-2000
|
2000-20000
|
Таблица 2. Классификация химических соединений по токсичности, принятая в СЭВ.
| Показатель
|
Классы токсичности веществ
|
| I
чрезвычайно токсичные
|
II
высокотоксичные
|
III
умеренно токсичные
|
IV
малотоксичные
|
| LD50
(мг/кг)
Введение в желудок
|
<15
|
15-150
|
151-5000
|
>5000
|
| LD50
(мг/кг)
Нанесение на кожу
|
<100
|
100-500
|
501-2500
|
>2500
|
| CL50
(мг/м3
)
Ингаляционное воздействие
|
<500
|
500-5000
|
5001-50000
|
>50000
|
Таблица 3. Классификация химических соединений по токсичности, принятая в КНР.
| Показатель
|
Классы токсичности веществ
|
| I
чрезвычайно опасные
|
II
высокоопасные
|
III
умеренно опасные
|
IV
малоопасные
|
| LD50
(мг/кг)
Введение в желудок
|
<25
|
25-500
|
500-5000
|
>5000
|
| LD50
(мг/кг)
Нанесение на кожу
|
<100
|
100-500
|
500-2500
|
>2500
|
| CL50
(мг/м3
)
Ингаляционное воздействие
|
<200
|
200-2000
|
2000-20000
|
>20000
|
В приведенных классификациях видно различие в выборе границ классов. В качестве примера укажем на то, что к малоопасным веществам при введении в желудок ФРГ относит токсиканты (пестициды), для которых LD50
более 1000 мг/кг, Греция – более 2000 мг/кг, ВОЗ – более 4000 мг/кг, а отечественный ГОСТ 12.1.007-76 – более 5000 мг/кг, то есть крайние значения LD50
в соответствии с этими классификациями различаются более чем в 2-5 раз [15].
Следует отметить, что рассмотренные классификации имеют ряд недостатков, главным из которых является произвольность выбора границ классов по среднесмертельным уровням LD50
.
Таким образом, проблема классификации химических соединений по показателю токсичности является не только проблемой современной теоретической и практической химии, но и немаловажной проблемой международного и экономического масштаба. В данной дипломной работе производится попытка создания универсальной научно обоснованной классификации химических по показателю токсичности LD50
соединений.
Учитывая вышеописанные факты, следует выделить в качестве одной из немаловажных задач данной работы — создание универсальной, математически обоснованной классификации химических веществ по показателю токсичности.
3. Разработка моделей прогнозирования токсикологических свойств химических веществ
Развитие различных отраслей промышленности, особенно химической, использование химических удобрений и пестицидов в сельском хозяйстве привели к постепенному загрязнению окружающей среды множеством химических веществ, которые в огромном количестве попадают в почву, воду и воздушную среду, где преобразуются в еще более токсичные продукты, вызывая отравления и различные заболевания, в том числе онкогенные. В целях предупреждения отрицательных последствий химизации народного хозяйства в различных странах сложились или создаются системы предупредительных мероприятий, среди которых одним из главных является токсикологическая оценка химических веществ и композиций, включая их предварительный отбор для последующего производства и применения. По данным ВОЗ в повседневном использовании, включая фармацевтические средства и пестициды, находятся более 60 тысяч опасных химических веществ. Проблемы предупреждения загрязнения окружающей среды вышли за рамки национальных границ и во многих случаях приобрели глобальный характер. Поэтому, поиск новых высокоэффективных и безопасных для человека и окружающей среды химических веществ является важнейшей проблемой мировой науки. Значительное место в этой проблеме занимает задача прогноза параметров токсичности (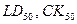 и др.) органических соединений, необходимость практического решения которой тесно связана со следующими обстоятельствами: и др.) органических соединений, необходимость практического решения которой тесно связана со следующими обстоятельствами:
· На этапе разработки и эксплуатации технологических процессов – с отставанием и неполнотой обоснования санитарно-гигиенических нормативов на используемое сырье, полупродукты, продукты и отходы (при использовании традиционных методов нормирования химических веществ и существующих мощностях токсикологических лабораторий необходимой токсикологической оценке и гигиеническому регламентированию подвергается не более 10% новых химических веществ;
· На этапе поисковых исследований (синтеза и биологических испытаний) – с необходимостью возможно более ранней оценки токсичности новых химикатов с целью дополнительной фильтрации токсичных целевых соединений и полупродуктов.
В связи с этим важное значение приобретает разработка альтернативных принципов нормирования и методологии ускоренной токсикологической оценки новых химических веществ, что позволит существенно сократить объем и время для экспериментального обоснования гигиенических и токсикологических нормативов. Поэтому, применение химических веществ требует тщательной проверки их безопасности для людей и окружающей среды и связано с использованием большого числа дорогостоящих тестов, альтернативой которым является математическое моделирование характера процесса взаимодействия химического вещества и живых организмов с использованием фактографических банков данных по показателям токсичности.
Таким образом, токсикометрия занимает значительное место в принятии радикальных решений по профилактике неблагоприятных воздействий химических веществ в окружающей среде. На стадии синтеза новых соединений и композиций она позволяет осуществлять целенаправленный отбор менее токсичных и опасных соединений, используя для этого целый набор качественных и количественных критериев. Широкое использование при таком отборе математических методов, компьютерных технологий и фактографических банков данных позволяет отсеивать заведомо неактивные или высокотоксичные вещества, тем самым значительно сокращая сроки создания физиологически активных соединений с заданными токсикологическими свойствами.
Математические модели прогноза токсичности.
Теоретической базой для построения моделей и развития расчетных методов определения токсичности является объективно существующая связь между токсическим действием вещества, его физическими свойствами и химической структурой. Из-за отсутствия в большинстве случаев адекватных теоретических представлений о механизмах биологического действия, из-за сложности процессов, происходящих с веществом в живых системах, широкое применение находят эмпирические закономерности, устанавливающие связь между строением молекул их физико-химическими и биологическими характеристиками. В данной работе будут исследоваться эмпирические обобщения в форме современных методов и моделей многомерной регрессии, а также теории распознавания образов. В качестве информационной поддержки исследуемых моделей будет использован фактографический банк данных по токсичности органических молекул объемом в 4624 соединений различных структурно-химических классов. Предсказание 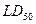 предполагается осуществлять в два этапа. На первом этапе должен осуществляться качественный прогноз, позволяющий определить класс токсичности или опасности вещества, что является весьма актуальной задачей, так как во многих химических исследованиях нет необходимости в строгой оценке параметров токсичности и достаточно знать классы опасности веществ. На втором этапе в каждом из классов токсичности нужно построить оптимальные регрессионные зависимости и по ним осуществлять количественный прогноз. предполагается осуществлять в два этапа. На первом этапе должен осуществляться качественный прогноз, позволяющий определить класс токсичности или опасности вещества, что является весьма актуальной задачей, так как во многих химических исследованиях нет необходимости в строгой оценке параметров токсичности и достаточно знать классы опасности веществ. На втором этапе в каждом из классов токсичности нужно построить оптимальные регрессионные зависимости и по ним осуществлять количественный прогноз.
Прогноз класса токсичности предполагается осуществлять на основе моделей и алгоритмов распознавания образов и теории статистических решений.
Количественный прогноз предполагается осуществлять на основе неаддитивных моделей с использованием понятия о парциальных вкладах структурных элементов.
В рамках данной задачи необходимо произвести исследование математических подходов прогноза токсикологических параметров, а также сравнение полученных результатов с результатами существующих коммерческих пакетов прогнозирования токсикологических свойств.
4. Система компьютерной поддержки.
Необходимо разработать автоматизированную информационно-поисковую систему, оснащенную математическими процедурами статистического моделирования токсикологических свойств химических веществ, состоящую из:
· Подсистемы поддержки профессиональных структурно-химических баз данных и знаний;
· Подсистемы прогнозирования тосикологических свойств органических молекул с учетом или без учета их физико-химических параметров. Она позволит создавать обучающие и экзаменационные выборки из баз данных, задавать или выбирать из меню различные описания химической структуры или иных признаков, выбирать различные модели статистической обработки данных для построения решений о принадлежности молекул к тому или иному классу токсичности, а также структурно-аддитивные и неаддитивные математические модели, которые используются для нахождения количественных корреляций структура – свойства.
Другими словами компьютерная система позволит осуществлять прогноз токсикологических параметров веществ с использованием моделей теории распознавания образов и кусочно-линейных регрессионных моделей, где интервалами линейности являются классы опасности химических соединений.
То, есть, необходимо создать компьютерную информационно-поисковую систему, которая даст возможность в режиме диалога вести оперативный прогноз токсикологических показателей, проверять на больших выборках гипотезы о связи структуры веществ с их биологическим действием, а также анализировать сравнительную информативную ценность различных групп факторов при изучении механизмов взаимодействия веществ с живым организмом. Такая система позволит повысить достоверность получаемых научных результатов и поможет существенно снизить трудоемкость исследовательских работ за счет качественно нового их уровня.
Научный задел.
Разработан математический подход классификации химических веществ по степени токсичности в острых опытах. Проведена апробация данного подхода на большом экспериментальном материале и установлены научно обоснованные границы классов опасности химических соединений. Разработана подсистема поддержки профессиональных структурно-химических баз данных и знаний. В ходе выполнения проекта в компьютерную систему нужно добавить подсистему расчета токсикологический параметров.
Таким образом, целью
данной работы является создание универсальной масштабируемой компьютерной системы, предназначенной для применения на практике алгоритмов поиска и анализа отношений "структура-активность". Такая система должна поддерживать как возможности информационного поиска и навигации, так и построения баз знаний на основе имеющихся данных. Также система должна быть открытой, расширяемой и максимально гибкой, с возможностью добавления новых возможностей.
В соответствии с целью поставлены следующие задачи:
1) Разработка математически обоснованной универсальной классификации химических соединений по показателю токсичности;
2) Разработка алгоритмов и методов для качественного прогнозирования принадлежности химического соединения к заданному классу токсичности.
3) Разработка и апробация моделей для количественного прогноза показателя токсичности LD50
;
4) Разработка и использование моделей для предсказания токсичности по липофильности;
5) Создание универсальной масштабируемой системы компьютерной поддержки, которая должна включать в себя:
¾ иерархию классов, обеспечивающих гибкость и универсальность в настройке и расширении приложения (framework):
¾ графический редактор структурных формул химических соединений;
¾ подсистему моделирования;
¾ подсистему хранения и информационного поиска данных;
Глава 2
Математическая модель классификации химических соединений по их различным свойствам
Известно, что в организованном сообществе элементы распределены в соответствии с гиперболическим законом, то есть:
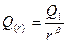 , (3.2.I) , (3.2.I)
где Q1
— количество элементов в первом классе,r — ранг класса (r = 1…n), Q(
r
)
— количество элементов в данном классе.
Для r=1, 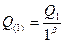 . (3.2.II) . (3.2.II)
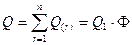 (3.2.III) , где Q — количество элементов сообщества, (3.2.III) , где Q — количество элементов сообщества,
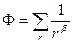 (3.2.IV). (3.2.IV).
Это уравнение дает общее решение по разбиению множества из Q элементов на n классов. Отсюда необходимо найти b.
По формуле Шеннона: 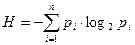 (3.2.V), где H — энтропия информации, pi
— вероятность попадания Qi
элементов множества Q в данный класс i, или (3.2.V), где H — энтропия информации, pi
— вероятность попадания Qi
элементов множества Q в данный класс i, или
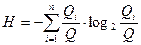 (3.2.VI). (3.2.VI).
Предельные значения энтропии информации равны 0 и Hmax
. Hmax
рассчитывается по формуле Хартли: Hmax
= log2
(n).
По принципу структурной гармонии Шеннона получаем обобщенное золотое сечение:
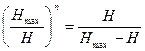 (3.2.VII), или (3.2.VII), или
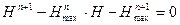 (3.2.VIII). (3.2.VIII).
Отсюда найдем H, как положительный действительный корень (по условию) полинома n+1 степени.
Подставляя (3.2.I) в формулу (3.2.VI), зная значение H, имеем:
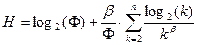 (3.2.IX). (3.2.IX).
Значение b, положительно определенное по условию, вычисляется из (3.2.IX) одним из численных методов решения уравнений. Далее, из (3.2.III) вычисляется значение Ф. После этого, подставляя Ф в (3.2.I), получаем количество элементов в каждом классе.
Для получения пределов значений показателя, по которому организовано (упорядочено) семейство, необходимо взять значения этого показателя для первого и последнего элемента каждого класса.
Регрессионные модели и их характеристики
Уравнение линейной регрессии имеет вид: y = a + bx + e [2].
Построение уравнения регрессии сводится к оценке ее параметров. Для оценки параметров регрессий, линейных по параметрам, используют метод наименьших квадратов (МНК). МНК позволяет получить такие оценки параметров, при которых сумма квадратов отклонений фактических значений результативного признака y от теоретических ŷx
минимальна, то есть:
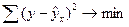 . .
Для линейных и нелинейных уравнений, приводимых к линейным, решается следующая система относительно a и b:
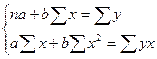 . .
Из этой системы следуют формулы:
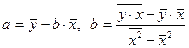 . .
Для расчета множественной линейной регрессии данные представляются в матричной форме [6]:
Y = Xb + e,
или
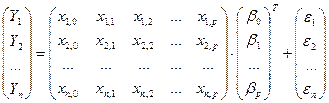 , ,
где матрица X называется регрессионной матрицей, вектор b — неизвестные параметры, подлежащие оцениванию, а столбец e — ошибки.
Пользуясь МНК, имеем:
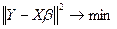 . .
В результате получаем выражение для оценки вектора b:
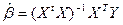 . .
Соответственно, появляется модель, связывающая экспериментальные данные:
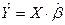 . .
Как для моделей парной, так и для множественной регрессии справедливы статистические оценки, описанные в таблице 7 [2]:
Таблица 7
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| Полная дисперсия
|
TSS
|

|
Общая сумма квадратов отклонений зависимой переменной от ее выборочного значения
|
| Часть дисперсии, необъясненная регрессией
|
ESS
|
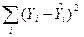
|
Необъясненная сумма квадратов отклонений
|
| Часть дисперсии, объясненная регрессией
|
RSS
|
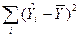
|
Объясненная сумма квадратов отклонений
|
| Коэффициент детерминации
|
R2
|
RSS/TSS
|
—
|
| F-статистика
(критерий Фишера)
|
F
|
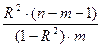
|
Оценка качества уравнения регрессии. Состоит в проверке гипотезы H0
о статистической незначимости уравнения регрессии. Для этого выполняется сравнение фактического F (где n – число единиц совокупности, m — число параметров при переменных x) и табличного (критического) Fтабл
. Fтабл
— это максимально возможное значение критерия под влиянием случайных факторов при данных степенях свободы и уровне значимости α. Уровень значимости α — вероятность отвергнуть правильную гипотезу при условии, что она верна. Обычно α принимается равным 0,05 или 0,01.
Если Fтабл
< F, то H0
— гипотеза о случайной природе оцениваемых характеристик отклоняется и признается их статистическая значимость и надежность. Иначе —
|
Продолжение таблицы 7
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| |
|
|
признается статистическая незначимость, ненадежность уравнения регрессии.
|
| Средняя ошибка аппроксимации
|
|
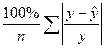
|
Среднее отклонение расчетных данных от фактических
|
Для расчета доверительных интервалов параметров линейной регрессии применяются статистически оценки, приведенные в таблице 8:
Таблица 8
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| Случайная ошибка параметра a линейной регрессии
|
ma
|
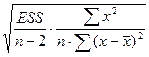
|
—
|
| Случайная ошибка параметра b линейной регрессии
|
mb
|
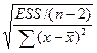
|
—
|
| t-критерий Стьюдента для параметра а
|
ta
|
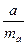
|
Рассчитывается для оценки статистической значимости коэффициентов регрессии. Выдвигается гипотеза H0
о случайной природе показателей, то есть о незначимом их отличии от нуля. Сравнивая фактическое и табличное (критическое) значения для заданного уровня значимости, принимаем или отвергаем выдвинутую гипотезу: если
|
| t-критерий Стьюдента для параметра b
|
tb
|
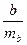
|
Продолжение таблицы 8
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| |
|
|
tтабл
> tфакт
, то H0
отклоняется, то есть a и b не случайно отличаются от нуля и сформировались под воздействием систематически действующего фактора x, иначе — природа формирования случайна.
|
| Доверительные интервалы параметров линейной регрессии
|

|
a- tтабл
ma
|
Если в границы доверительного интервала попадает ноль, то есть нижняя граница отрицательна, а верхняя положительна, то оцениваемый параметр принимается нулевым, так как он не может принимать и положительное, и отрицательное значение.
|
| 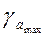
|
a+ tтабл
ma
|
| 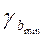
|
b- tтабл
mb
|
| 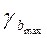
|
b+ tтабл
mb
|
Во множественной регрессии для нахождения доверительных интервалов справедливы формулы, описанные в таблице 9:
Таблица 9
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| Дисперсия остатков регрессии
|
s2
|
ESS/(n-k)
|
n — число единиц совокупности, k — число неизвестных параметров.
|
| Дисперсия i-го коэффициента регрессии
|
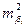
|
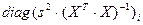
|
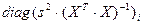
— i-й элемент диагонали ковариационной матрицы
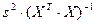
|
Продолжение таблицы 9
| Название характеристики
|
Обозначение
|
Формула
|
Описание
|
| Доверительный интервал i-го параметра множественной регрессии
|
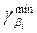
|
|
Свойства аналогичны свойствам доверительных интервалов для парной регрессии. Табличное
|
| |
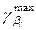
|
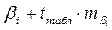
|
значение t-критерия Стьюдента выбирается для n-k степеней свободы.
|
Модель качественного прогноза
Прогноз класса токсичности осуществляется на основе моделей и алгоритмов распознавания образов и теории статистических решений. Мы рассматривали задачу распознавания образов применительно к случаю двух классов. Это весьма распространенный случай, так как при любом другом числе классов последовательным разбиением на два класса можно построить разделение и на произвольное число k классов. Для этого достаточно провести k разбиений по принципу: отделить элементы первого класса от смеси остальных, затем элементы второго класса от остальных и т. д.
Обозначим через 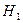 соответствующий класс токсичности. Будем рассматривать объекты обучающей выборки, входящие в , как положительные примеры класса соответствующий класс токсичности. Будем рассматривать объекты обучающей выборки, входящие в , как положительные примеры класса 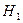 , а объекты, не входящие в , — как контрпримеры или отрицательные объекты класса , множество которых мы обозначим через , а объекты, не входящие в , — как контрпримеры или отрицательные объекты класса , множество которых мы обозначим через 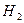 . Запишем бинарный вектор наблюдений X в виде . Запишем бинарный вектор наблюдений X в виде 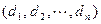 , где , где 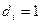 или 0 в зависимости от того, присутствует или отсутствует i-й фрагмент структуры в описании соединения. Обозначим через или 0 в зависимости от того, присутствует или отсутствует i-й фрагмент структуры в описании соединения. Обозначим через 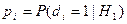 и и 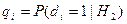 вероятности появления i-го дескриптора в классах и соответственно. вероятности появления i-го дескриптора в классах и соответственно.
В предположении условной независимости можно записать условные плотности распределения вероятностей в каждом классе в виде произведения вероятностей для компонент вектора наблюдений.
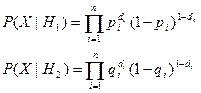
Отношение правдоподобия при этом определяется выражением
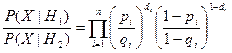 .
.
Прологарифмировав это отношения и приведя подобные члены, получим байесовскую решающую функцию
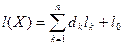 , ,
где 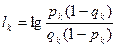 — информационный вес k-го дескриптора, а — информационный вес k-го дескриптора, а
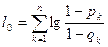 — константа. — константа.
Байесовское решающее правило, минимизирующее среднюю вероятность ошибки, согласно [5], записывается следующим образом:
если 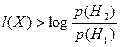 , то , то 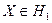 , иначе , иначе 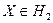 . .
При выводе решающего правила мы исходили из того, что потери при правильной классификации равны нулю, а при ошибочной единице. При построении систем распознавания возможны такие ситуации, когда априорные вероятности появления объектов соответствующих классов 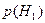 и и 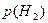 неизвестны. Применительно к этой ситуации рационально использовать минимаксный критерий, который минимизирует максимально возможное значение среднего риска. Показано [16], что минимаксное правило представляет собой специальное правило Байеса для наименее благоприятных априорных вероятностей. В этом случае решающая граница выбирается так, чтобы обеспечить равенство ошибок первого и второго рода, которые соответственно равны: неизвестны. Применительно к этой ситуации рационально использовать минимаксный критерий, который минимизирует максимально возможное значение среднего риска. Показано [16], что минимаксное правило представляет собой специальное правило Байеса для наименее благоприятных априорных вероятностей. В этом случае решающая граница выбирается так, чтобы обеспечить равенство ошибок первого и второго рода, которые соответственно равны:
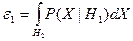 и и 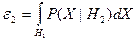 . .
Оценка величин pi
и qi
осуществляется по конечному числу выборочных представителей образов в соответствующих классах:
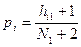 , , 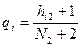 , ,
где 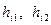 — числа встречаемости i-го дескриптора в первом и втором классах, а — числа встречаемости i-го дескриптора в первом и втором классах, а 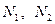 — объемы выборок в этих классах. — объемы выборок в этих классах.
Отнесение химического соединения к соответствующему классу токсичности производилось в дипломном проекте по значениям 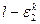 , где , где 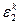 — ошибка второго рода для k-го класса в зависимости от отношения правдоподобия l, а значение k, на котором достигается — ошибка второго рода для k-го класса в зависимости от отношения правдоподобия l, а значение k, на котором достигается 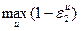 , и является номером класса опасности. , и является номером класса опасности.
Модель количественного прогноза
Количественный прогноз осуществлялся на основе неаддитивных моделей с использованием понятия о парциальных вкладах структурных элементов (дескрипторов). Используемые модели параметров, входящих в сртуктурно-неаддитивные модели имеют вид
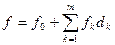 , ,
где fk
— парциальный вклад k-х дескрипторов в параметр f, dk
— доля k-х структурных элементов в молекуле
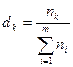 . .
В нашем случае в качестве параметра f использовался нормированный показатель токсичности
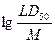 , ,
где M есть молекулярная масса молекулы. В каждом из классов опасности строились оптимальные регрессионные уравнения, в которых величины fk
определялись исходя из экспериментальных данных устойчивым методом наименьших квадратов, а также при помощи сингулярного разложения матрицы (п.4.3.1).
Алгоритм сингулярного разложения матрицы и приближенного решения алгебраических систем линейных уравнений
При описании различных моделей могут возникать системы линейных алгебраических уравнений с прямоугольными и вырожденными квадратными матрицами. Для систем линейных алгебраических уравнений, не обладающих решением с классической точки зрения, вводят понятие обобщенного решения [9]. Под обобщенным решением (псевдорешением) системы линейных алгебраических уравнений
Ах = b,
(4.3.1.I)
где А
– матрица с размерами m x n, b –
заданный вектор, x –
искомый вектор, понимают вектор u
, удовлетворяющий условию
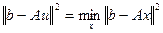 , (4.3.1.II) , (4.3.1.II)
где || || - евклидова норма.
Если система (4.3.1.I) имеет классическое решение, то оно совпадает с обобщенным, и при этом 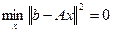 . Однако, нахождение векторов, минимизирующих функционал невязки . Однако, нахождение векторов, минимизирующих функционал невязки 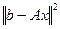 , имеет смысл и в отсутствии классического решения системы (4.3.1.I). Поэтому введение определения обобщенного решения существенно расширяет понятие искомого решения системы (4.3.1.I). , имеет смысл и в отсутствии классического решения системы (4.3.1.I). Поэтому введение определения обобщенного решения существенно расширяет понятие искомого решения системы (4.3.1.I).
В работе Воеводина В.В. "Линейная алгебра" доказано, что для системы (4.3.1.I) всегда существует множество псевдорешений, а если рассмотреть так называемое нормальное псевдорешение, то есть решение с минимальной евклидовой нормой, то оно еще и единственно.
Для решения системы (4.3.1.I) в дипломной работе было использовано специальное представление матрицы, называемое сингулярным разложением. Известно, что любую действительную матрицу с размерами m x n можно представить в виде
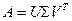 , (4.3.1.III) , (4.3.1.III)
где матрица U (m x m) сформирована из m ортонормированных собственных векторов матрицы AAT
, матрица V (n x n) — из n ортонормированных собственных векторов матрицы AT
A, матрица S с размерами m x n имеет вид 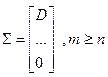 , или , или 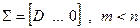 , при , при 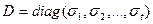 [9]. [9].
Диагональные элементы si
являются неотрицательными значениями квадратных корней из общих собственных значений матриц AAT
и AT
A и называются сингулярными числами матрицы А. Если сингулярные числа упорядочены, то такое разложение называется сингулярным разложением матрицы А.
Зная сингулярное разложение, можно сразу выписать решение системы (4.3.1.I):
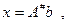 где A#
=VS#
UT
называется псевдообратной к А матрицей. где A#
=VS#
UT
называется псевдообратной к А матрицей.
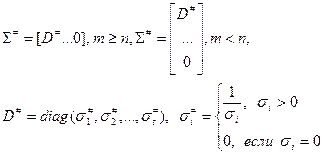 . .
Преобразование прямоугольной матрицы А к двухдиагональному виду [11], [14]
Первым этапом нахождения сингулярного разложения матрицы А является ее численное приведение при помощи преобразований Хаусхолдера к двухдиагональному виду. Рассмотрим это преобразование.
Умножая слева и справа исходную матрицу А соответственно на специально подбираемые матрицы отражения P(
k
)
и Q(
k
)
, приходят к верхней двухдиагональной форме
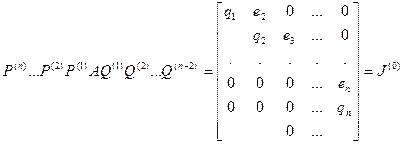 . .
Процесс преобразования осуществляется по формулам
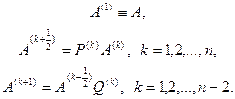
Матрицы отражения P(
k
)
и Q(
k
)
следует выбирать так, чтобы были выполнены условия

В этом случае матрицы P(
k
)
, Q(
k
)
будут иметь вид
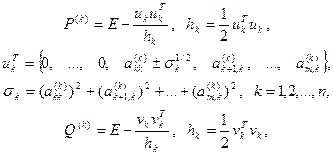
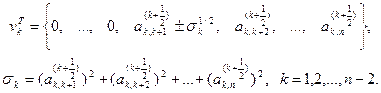
Знак перед 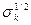 в выражениях для в выражениях для 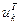 и и 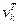 следует выбирать таким же, как и знаки следует выбирать таким же, как и знаки 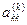 и и 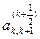 соответственно. соответственно.
Окончательно введя обозначения
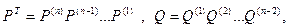
можно записать 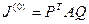 . .
Здесь P и Q — ортогональные матрицы. При таком преобразовании сингулярные числа матрицы J(0)
совпадают с сингулярными числами матрицы А.
Сингулярное разложение двухдиагональной матрицы
Следуя [17], изложим алгоритм сингулярного разложения двухдиагональной матрицы. С помощью так называемого QR-метода можно привести двухдиагональную матрицу J(0)
к диагональной форме D, так что выполняется последовательность преобразований
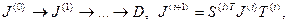 (4.3.1.IV) (4.3.1.IV)
где S(
i
)
и T(
i
)
— ортогональные матрицы, которые выбирают так, чтобы J(
i
+1)
сохраняли свою двухдиагональную форму, а симметричная трехдиагональная матрица J(
i
)
T
J(
i
)
стремилась к диагональному виду.
Для удобства опустим индексы и введем следующие обозначения:
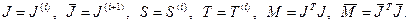
Переход  осуществляется с помощью последовательности преобразований вращения. Таким образом, осуществляется с помощью последовательности преобразований вращения. Таким образом,
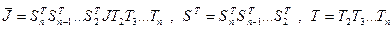 . (4.3.1.V) . (4.3.1.V)
Здесь Sk
и Tk
— элементарные матрицы вращения вида
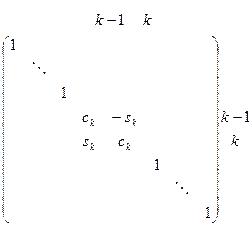 , ,
причем 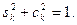 Для общего случая коэффициенты c и s вычисляются по формулам Гивенса Для общего случая коэффициенты c и s вычисляются по формулам Гивенса
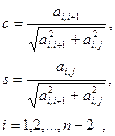
где ai
,
j
— вытесняемый элемент.
Очевидно, что умножение справа на матрицу вращения изменяет лишь (k-1) и k столбцы матрицы, а умножение слева на матрицу вращения — лишь (k-1) и k строки. Формулы преобразования для столбцов имеют вид
 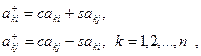
для строк
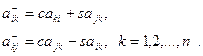
Коэффициенты c2
, s2
матрицы T2
оставим пока неопределенными, в то время как остальные коэффициенты ck
, sk
будем выбирать так, чтобы матрица 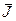 имела ту же форму, что и J. Следовательно, матрица T2
не аннулирует ни одного элемента, но добавляет элемент J21
, матрица S2
аннулирует J21
, но добавляет J13
, матрица T3
аннулирует J13
, но добавляет J32
и т. д. и окончательно матрица имела ту же форму, что и J. Следовательно, матрица T2
не аннулирует ни одного элемента, но добавляет элемент J21
, матрица S2
аннулирует J21
, но добавляет J13
, матрица T3
аннулирует J13
, но добавляет J32
и т. д. и окончательно матрица 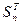 аннулирует Jn
,
n
-1
и ничего не добавляет. аннулирует Jn
,
n
-1
и ничего не добавляет.
При этом справедливы следующие соотношения:
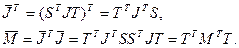
Таким образом, ортогональное преобразование (4.3.1.V) эквивалентно преобразованию подобия симметричной трехдиагональной матрицы
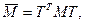 (4.3.1.VI) (4.3.1.VI)
где матрица 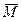 будет также трехдиагональной, поэтому матрицу T2
, которая пока еще не определена, необходимо выбирать так, чтобы преобразование будет также трехдиагональной, поэтому матрицу T2
, которая пока еще не определена, необходимо выбирать так, чтобы преобразование 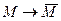 было QR-преобразованием со сдвигом, равным s. было QR-преобразованием со сдвигом, равным s.
Обычно QR-алгоритм можно записать в следующем виде:
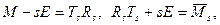 (4.3.1.VII) (4.3.1.VII)
где 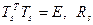 — верхняя треугольная матрица. Но не обязательно выполнять вычисления по формулам (4.3.1.VII). Сдвиг можно осуществлять и неявным образом. Доказано, например, в работе [17], что определенным выбором T2
можно добиться того, чтобы преобразование (4.3.1.VI) было эквивалентно QR-преобразованию для M с заданным сдвигом s. — верхняя треугольная матрица. Но не обязательно выполнять вычисления по формулам (4.3.1.VII). Сдвиг можно осуществлять и неявным образом. Доказано, например, в работе [17], что определенным выбором T2
можно добиться того, чтобы преобразование (4.3.1.VI) было эквивалентно QR-преобразованию для M с заданным сдвигом s.
Пусть T и Ts
ортогональные матрицы такие, что выполняются следующие условия:
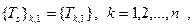
то есть элементы первого столбца Ts
равны элементам первого столбца T и
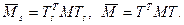
Тогда, если поддиагональные элементы матрицы M ненулевые, то матрица 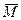 связана с связана с 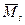 следующим образом: следующим образом:
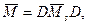
где в — диагональная матрица, элементы которой равны ±1.
Следовательно, задача заключается лишь в том, чтобы первый столбец матрицы T выбрать равным первому столбцу матрицы Ts
. Далее, имеем
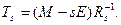
Учитывая тот факт, что обратная матрица к верхней треугольной будет также верхней треугольной, можно сделать вывод, что первый столбец искомой матрицы Ts
будет пропорционален первому столбцу матрицы M — sE.
Таким образом, матрица T будет матрицей QR-преобразования со сдвигом s, если ее первый столбец будет пропорционален первому столбцу матрицы M — sE. А так как T = T2
T3
…Tn
, окончательно приходим к выводу, что T2
в (4.3.1.V) необходимо выбирать так, чтобы ее первый столбец был пропорционален первому столбцу M — sE. В этом случае преобразование (4.3.1.V) будет эквивалентно QR-преобразованию со сдвигом s для матрицы M. Параметр сдвига s выбирается равным собственному значению нижнего минора матрицы M,
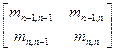 , ,
которое ближе к mn
,
n
. При таком выборе параметра метод обладает глобальной и почти всегда кубической сходимостью [17].
Таким образом, в результате преобразования (4.3.1.IV), (4.3.1.V) сингулярное разложение для матрицы J(0)
будет иметь вид
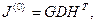
где G и H — ортогональные матрицы.
|