МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ
РЕСПУБЛИКИ КАЗАХСТАН
КАЗАХСКИЙ НАЦИОНАЛЬНЫЙ УНИВЕРСИТЕТ им.АЛЬ- ФАРАБИ
Химический факультет
Кафердра неорганической химии
Выпускная работа
ПОЛУЧЕНИЕ ПОЛИМЕРНЫХ ФОСФАТОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ И ИЗУЧЕНИЕ ИХ СОРБЦИОННЫХ СВОЙСТВ
.
Исполнитель
студент 4 курса : Калинина А.В.
Научный руководитель:
д.х.н., профессор
«
___
» июня 2010г. Куанышева Г.С.
Нормоконтролер: Рыскалиева Р.Г.
Допущена к защите «
___
» июня 2010г.
Зав.кафедрой неорганической химии
д.х.н., профессор Абрамова Г.В.
Алматы 2010г
Р Е Ф Е Р А Т
Выпускная работа состоит из введения, трех разделов , выводов , списка использованных источников, состоящего из 40 наименований. Работа изложена на ___ страниц машинописного текста, включает __ рисунков и ___ таблиц.
Перечень ключевых слов: дифосфаты, сорбция, ионный обмен, комплексообразование ...
Работа посвящена проблеме изучения и исследования полимерных фосфатов в качестве новых неорганических сорбентов и обнаружению факторов влиящих на их сорбционные свойства. Также поставлена задача установления механизма протекающего процесса.
Во время проведения работы использованы следующие аналитические методы: фотоколометрический метод, рентгенофазовый и ИК-спектроскопический и химический анализ.
Содержание
Введение . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1. ЛИТЕРАТУРНЫЙ ОБЗОР . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.1. Краткие сведения о конденсированных фосфатах. . . . . . . . . . . . . . . . . . 6
1.2. Общие сведения о неорганических ионитах. . . . . . . . . . . . . . . . . . . . . . . . 9
1.3.
Селективные и комплексообразующие иониты . . . . . . . . . . . . . . . . . .
.
15
1.4. Основные характеристики ионообменников. . . . . . . . . . . . . . . . . . . . . . 19
1.5. Ионообменные процессы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.1. Характеристика исходных веществ. Методика химических и физико-химических методов исследования. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.2. Синтез дифосфатов Со,
Ni
и
Fe
и их характеристика. . . . . . . . . . . . . . . 26
2
.
3. Идентификация синтезированных фосфатов Со,
Ni
и
Fe
и определение
их свойств. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
2.4. Исследование сорбицонных свойств . . . . . . . . . . . . . . . . . . . . . . . . . . . . .38
2.5. Химизм ионного процесса. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3. ВЫВОДЫ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
СПИСОК ИСПОЛЬЗУЕМОЙ ЛИТЕРАТУРЫ . . . . . . . . . . . . . . . . . . . . . . . 45
Введение
Казахстан занимает ведущее место в стране по запасам фосфоритов. Фосфатное сырье республики – один из главных источников фосфора для производства фосфорных удобрений кормовых фосфатов, фосфорно-кислых солей, моющих средств и других ценных продуктов, применяемых в сельском хозяйстве технике, биологии, медицине и т.п.
Исследование адсорбции, комплексообразования и ионного обмена в растворах фосфатов позволяет изучать химизм превращений фосфорных удобрений в почве и изыскивать пути повышения их эффективности. Работы в области химии и технологии фосфатов различного назначения ведутся также в Казахском Национальном университете им. аль-Фараби на кафедре неорганической химии. Это прежде всего создание технологии удобрений на базе актюбинских фосфоритов совместная переработка фосфатного и калийного сырья с получением продуктов типа ренанита и полифосфатов.
В последние годы неорганические ионообменные материалы привлекают все большее внимание, главным образом вследствие их способности удовлетворять ряду требований, предъявляемых современной техникой; к ним относятся, в частности, устойчивость при повышенных температурах и достаточно интенсивном радиоактивном излучении. Как известно, существующие многочисленные органические ионообменные смолы, применение которых хорошо разработано, неустойчивы в указанных условиях.
Существует огромное множество неорганических веществ, которые, как кажется с первого взгляда, могли бы выполнять функции ионообменников в определенных экстремальных условиях. К ним относится прежде всего многочисленные природные минералы с силикатным скелетом. Включающие в свой состав наряду с такими типичными для них катионами, как алюминий, кальций, железо, магний и т.д., катионы щелочных металлов, чаще всего натрия, и калия, наиболее способные к ионному обмену. Не меньшее значение имеют силикаты, в которых способные к обмену ионы водорода находятся в форме гидроксильных групп или анионов.
Вопросы сорбции тяжелых металлов неорганическими сорбентами представляют самостоятельный интерес. Большое разнообразие сфер применение этого явления делает актуальным всестороннее изучение кинетики сорбции ионов с осадками различного типа. Среди многочисленных труднорастворимых соединений-сорбентов внимание исследователей постоянно привлекают фосфаты металлов, поскольку они обладают большей емкостью и избирательностью по отношению ко многим ионам. Сведения по сорбции ионов марганца (II) дифосфатами железа, цинка и кобальта в литературе весьма ограничены.Поэтому целью настоящей работы является восполнение этих данных: осуществить синтез дифосфатов Со, Zn, Ni, и Fe2+
изучить их ионообменную емкость.
1.
Литературный обзор.
1.1. Краткие сведения о конденсированных фосфатах.
Конденсированные фосфаты издавна были объектом многих исследований. Эти соединения многочисленны и существуют в виде специфических кристаллических солей и в виде аморфных стекол, последние являются смесью веществ. Все разновидности образованы в результате неоднократной конденсации тетраэдрических групп РО4
, которые через общие атомы кислорода могут иметь общие вершины с подобными же тетраэдрами.
О-
О-
||
-
О – Р – О – Р – О-
| |
О О
Простейшим представителем конденсированных фосфатов является дифосфат (пирофосфат-) анион, который образуется при конденсации двух ортофосфатных анионов. Одним из наиболее простых способов получения полимеризованных фосфатов является процесс поликонденсации. Это процесс состоит в усложнении фосфорных анионов за счет межмолекулярного выделения воды согласно по схеме:
О-
О-
О-
О-
| | | |
-
О – Р – ОН + НО – Р – О-
→ О – Р – О – Р – О-
+Н2
О
| | | |
О-
О-
О-
О-
В этом процессе теряются два отрицательных заряда и образуется новый анион и образуется новый анион Р2
О7
4-
. Тетраэдры РО4
в конденсированных фосфатах никогда не соединяются через общие ребро или грань, но каждый тетраэдр может иметь максимально три общие вершины с соседними тетраэдрами.
Конденсированные фосфаты разделяются на три основных типа:
1. Линейные фосфаты (полифосфаты), образующие бесконечный ряд солей, анионы которых состоят из РО4
-
тетраэдров, соединенных друг с другом атомами кислорода в неразветвленные цепи. Общий состав полифосфатов соответствует формуле МI
n+2
Pn
O3n+1
.
2. Циклические фосфаты (метафосфаты) состава МI
n
Pn
O3
n
, которые при n=(3,4) имеют кольцевую структуру;
3. Разветвленные фосфаты (ультрафосфаты) представляют собой комбинации колец и цепей с общей формулой аниона Pn
O3
n
+
m
.
Все конденсированные фосфаты можно представить в виде общей формулы Мn
Pn
O(5+
R
/2)
, где М – эквивалент катиона, R – молярное отношение оксида металла пентоксида фосфора, т.е. R = МеО/Р2
О5
, n – степень полимеризации. Величина R определяет вид конденсированного фосфата (линейных, циклических, разветвленных фосфатов).
Существуют несколько структурных блоков, с помощью которых можно представить структуру цепных разветвленных и кольцевых полимерных соединений. Наиболее распространенными являются:
О – М
|
– О – Р = О – концевая группа,
|
О – М
О – М
|
–
О – Р – О-
–
срединная группа,
| |
O
О-
|
–
О – Р – О-
–
точка разветвления.
| |
О
Вследствие трудностей разделения образующихся полимерных продуктов, каждая группа конденсированных фосфатов изучена недостаточно, хотя из практики известно, что некоторые из них, например, метафосфаты используются в качестве концентрированных удобрений.
Конденсированные фосфаты содержат атомы Р в полностью окисленном состоянии, поэтому химически они довольно устойчивы. Однако хорошо известна их гидролитическая неустойчивость и в определенных условиях все связи Р – О – Р в структуре могут быть разрушены. Конечным продуктами гидролиза являются дискретные ортофосфат – ионы, хотя ход и скорость гидролиза характерны для определенных конденсированных анионов и применяемых условий.
В зависимости от степени полимеризации n (степени конденсации, длины цепочки) полифосфаты подразделяются на низкомолекулярные фосфаты, где n<10, и высокомолекулярные (высокополимерные, длинноцепочечные), где n>50.
Фосфаты, содержащие более одного катиона, названы двойными, тройными и т.д. (в общем случае – разнокатионными), а в случае более сложного анионного состава – смешанными фосфатами.
Разнообразие состава возможных фосфатов двухвалентных металлов, характеризуемое, в частности, величиной замещенности фосфата, его гидратностью, степенью поликонденсации фосфатного аниона, природой катиона и т.д., предопределяет существенные различия в способах и конкретных условиях получения заданного фосфата. Следует отметить, что получение многих фосфатов поли валентных металлов, свободных от примеси других фосфатов, до настоящего времени в некоторых случаях химической промышленностью не реализовано. Многие фосфаты двухвалентных металлов, выпускаются химической промышленностью только квалификации «чистые» [1, 2].
Конденсированные фосфаты являются уникальным классом неорганических соединений, которые, подобно органическим веществам, образуют гомологические ряды олигомерных и полимерных производных, устойчивых не только в твердом состоянии, но и в водных растворах . Благодаря широкому спектру полезных для практики свойств, обусловленных полимерным строением, этот класс фосфатов давно привлекает внимание многочисленных исследователей.
Учеными, в частности, ведутся работы в области синтеза разнообразных типов конденсированных фосфатов: получены соединения с крупными кольцевыми анионами из 10 и I2 тетраэдров PO4
, цепочечные фосфаты, содержание до 5 тетраэдров PO4
, разнообразные кристаллические ультрафосфаты. Широко развиты синтез и исследованиие конденсированных фосфатов, в которых часть атомов кислорода замещена на атомы азота (циклофосфиматы, нитридофосфаты), серы (тиофосфаты) или галогена (фторфосфаты). Показано, что кольцевые анионы, в которых мостиковые атомы кислорода замещены на азот — циклофосфиматы, представляют собой новый обширный класс неорганических лигандов, обладающих высокой информационной лабильностью за счет подвижности связей фосфор – азот. Результаты исследований кристаллохимии конденсированных фосфатов позволили не только расшифровать строение практически всех типов полученных фосфатов, но в ряде случаев также объяснить их свойства и прогнозировать синтез тех или иных соединений. Успешно изучается механизм перестройки конденсированных фосфатов при повышенных температурах в кристаллах, стеклах и расплавах с использованием методов высокотемпературной рентгенография и Раман-спектроскопии. В последние годы появились новые области применения конденсированных фосфатов в качестве удобрений пролонгированного действия, материалов для квантовой электроники, волоконной оптики и других областей новой техники [3].
Известно, что конденсированные фосфаты образуют гомологические ряды соединений с циклической, линейной и разветвленной и структурами анионов. Однако в индивидуальном состоянии получить те или иные разновидности фосфатов очень сложно. До настоящего времени выделены и изучены лишь простейшие кольца из 3 и 4 тетраэдров РО4
, цепи из 2, 3 и 4 тетраэдров и некоторые формы длинноцепочечных полифосфатов, индивидуальные соединения с разветвленной структурой в кристаллическом виде вообще не были получены. Поэтому проблема синтеза новых соединений и химии конденсированных фосфатов по-прежнему остается актуальной.
Вследствие легкости перестройки фосфатных анионов по связям Р – О – Р в отличие от органических веществ в препаративной химии конденсированных фосфатов специфических и хорошо воспроизводимых приемов циклизации или наращивания длины цепи не найдено.
В связи с этим наиболее приемлемым является подход, в котором используется способность катиона влиять на формирование той или иной формы аниона в процессе образования или перестройки фосфатного полимера. Из множества вероятных конфигураций фосфатных анионов выбирается и стабилизируется та, которая в наибольшей степени удовлетворяет геометрическим и координационным требованиям катиона- металла, Тем самым важное значение приобретает установление корреляций между свойствами катионов (размером, зарядом, строением электронных оболочек) и строением образующихся соединений, что позволяет прогнозировать и получать те или иные типы фосфатных соединений.
В частности, соединения, выделенные в системах М2
О3
–Р2
О5
-Н2
О для трехвалентных металлов, и их комбинаций, со щелочными металлами, состоят из ди-, три-, тетра-, поли-, цикло- и ультрафосфатов разнообразных типов (табл. 1.) Кроме того, в них обнаружен новый тип соединений, содержащих одновременно два аниона разной степени конденсации - три- и циклотетрафосфат, каждое из которых характерно не для одного катиона-металла, а для группы, катионов а близкими ионными радиусами.
Таблица 1
Основные типы негидратированных конденсированных фосфатов трехвалентных металлов.
| Форма анионов |
Состав соединений |
Катины МІІІ
, с которыми получены соединения |
| Дифосфаты |
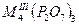
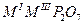
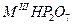
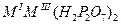
|
V, Cr, Fe, In
Al, Cr, Mn, Fe, Ga, In, Sc, Ho,Er, Tm-Lu
Mn, Fe, Tm-Lu
Al, Ga, I, Mn, Fe
|
| Трифосфаты |
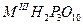
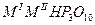
|
Al, Mn, Fe, Ga, In, Yb
Al, Mn, Fe, Ga, In, Pr - Lu
|
| Тетрафосфаты |
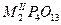 |
Bi, Eu |
| Разнолигандные фосфаты (три-циклотетрафосфаты) |
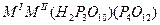 |
Al, Fe |
| Высокомолеулярные полифосфаты |
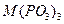
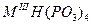
|
Все металлы III группы,
Mn, V, Fe, Cr, Bi
Sm – Eu, Bi
|
| Циклотетрафосфаты |
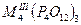
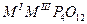
|
Al, Cr, Fe, р.з.э.
Al, Ga, In, р.з.э.
|
| Циклогексафосфаты |
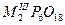 |
Al, Cr, Fe |
| Циклооктафосфаты |
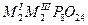 |
Al, Ga, In |
| Циклододекафосфаты |
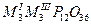 |
Al, Ga, V, Fe |
| Ультрафосфаты |
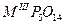 |
La – Lu, Bi |
Изучение свойств получаемых соединений позволило выявить более предпочтительные области их практического применения. Так, например, повышенная термическая устойчивость полифосфатов трехвалентных металлов при высоком содержание фосфора позволяет применять их в качестве сырьевых компонентов при производстве фосфатных стекол (вместо смесей оксидов металлов с легколетучими фосфорной кислотой или фосфатом аммония). Низкое концентрационное тушение люминесценции ионов Nd3+
, низкие пороги накачки и высокие квантовые выходы, характерные для высококонденсированных фосфатов неодима, обусловили использование кристаллических и стеклообразных ультрафосфатов, а также двойных полифосфатов неодима и щелочных металлов в квантовой электронике, в частности для изготовления миниатюрных лазерных устройств. Высокой каталитической активностью обладают фосфаты ванадия, применяемые в органическом синтезе. Перспективны в качество высокоэффективных фото- и катодолюминесцентных материалов конденсированные фосфаты европия и др.
Все это показывает, что конденсированные фосфаты представляют собой соединения с чрезвычайно богатой и сложной химией, Обладающие широким спектром свойств, что расширяет возможности применения фосфатов в различных областях техники [4, 6].
1.2. Общие сведения о неорганических ионитах.
Иониты нашли широкое применение в химической, медицинской, фармацевтической и других отраслях промышленности для умягчения и обессоливания воды, для селективного извлечения цепных металлов из различных растворов, для очистки сточных вод от неорганических и органических примесей, в химическом анализе, при получении веществ особой чистоты, в качестве катализаторов. А также в различных видах хроматографии.
Иониты твердые нерастворимые органические полимеры, в структуре которых содержатся функциональные (ионогенные) группы, способные к диссоциации и обмену подвижных ионов на ионы других соединений в растворе. Наряду с реакциями ионного обмена функциональные группы ионитов могут участвовать в реакциях комплексообразования и в окислительно-восстановительных процессах [7].
Многообразие неорганических ионитов практически неисчерпаемо. Иные из них естественного происхождения, но еще больше — искусственного. Среди изученных неорганических ионитов преобладают катиониты; анионитов известно совсем немного: апатит Са3
(Р04
)2
СаСlF, меркарбид [C6
Hg6
O2
]Cl2
·2H2
O, гидроксид циркония, недавно синтезированный ионит [ZnCr(OH)6
]Cl·2H2
O. Это обусловлено большим размером анионов, упаковка которых определяет структуру кристаллической решетки неорганических кристаллов. Имеется и много амфотерных ионитов, которые в зависимости от кислотности среды, а также от условий синтеза обладают либо катионо-, либо анионообменными свойствами.
У большей части неорганических ионитов трехмерная кристаллическая решетка, однако распространены и слоистые решетки: у некоторых цеолитов (гейландит, стильбит), у слюд, длинных минералов, урановых слюд (уранофосфатов, ураноарсенатов, уранованадатов), оксида графита, многих ферроцианидов, цирконилфосфата, меркарбида, многих солей поли-, кислот титана, ванадия, ниобия, молибдена, вольфрама и урана, фосфатов циркония и мышьяка.
Иониты с волокнистой структурой — это некоторые цеолиты (натролит, эдингтопит, сколецит, томсонит), длинный минерал аттапульгит (палыгорскит), кристаллы которого имеют экзотическую форму пустотелых трубочек.
Имеются и аморфные иониты: аморфные алюмосиликаты (пермутиты), силикагель, стекла. Структура многих гидроксидов в зависимости от условий синтеза может быть как кристаллической, так и аморфной.
Обычные (не пористые) ионные кристаллы способны к обмену ионов, сорбированных на поверхности, либо находящихся в приповерхностном слое кристалла. Однако при развитой поверхности кристаллического порошка обменная способность таких кристаллов может быть значительной. Вопросам ионообменной сорбции на поверхности ионных кристаллов уделялось много внимания в аналитической химии и радиохимии. Поверхностная ионообменная сорбция весьма важна и в химии коллоидов, так как структура и заряд определяющего устойчивость коллоидного раствора двойного электрического слоя на поверхности коллоидных частиц существенно зависит от состава раствора. Это подтверждено прямыми исследованиями коагуляции растворов гидрофобных коллоидов, смесями электролитов.
А также хорошо известно явление изоморфного замещения ионов в кристаллической решетке. Близкие по размерам катионы, образующие с данным анионом сходные кристаллические решетки, могут давать смешанные кристаллы, состав которых может меняться в определенном диапазоне. Изменение состава крупных кристаллов путем обмена их ионов с раствором — очень медленный процесс. Однако в ряд случаев при увеличении температуры и давления изоморфное замещение одного иона на другой значительно ускоряется. Это явление часто рассматривают как точный обмен, считая фиксированной группой образующие основу кристаллической решетки практически неподвижные анионы, а за обменные ионы катионы.
В изоморфном замещении, в отличие от обычного ионного обмена, участвуют лишь те катионы, структурные параметры которых соответствуют данной кристаллической решетке. Этот предельный случай характерной для ряда неорганических ионитов способности отделять друг от друга молекулы и ионы — разного размера, называемой молекулярно или, соответственно, ионно-ситовым действием. Оно обусловлено наличием в кристалле ионитов каналов и пор строго определенного размера, в которые проникают лишь те ионы и молекулы, эффективный диаметр которых не превышает эффективного сечения этих каналов. В результате сорбционного или обменно-сорбционного процесса удается «отсеять» друг от друга ионы и молекулы, различающиеся по эффективному диаметру. Это помогает выполнять трудно осуществимое другими способами разделение смесей близких по свойствам веществ, в том числе изомеров.
По распространенности в природе, многообразию свойств и применений особо выделяются иониты из группы алюмосиликатов: глины, слюды, цеолиты, полевые шпаты и т.п. Свойства цеолитов и глин далее будут рассмотрены специально.
Имеются иониты и среди других классов соединений. Прежде всего это многочисленные простые и сложные фосфаты и полифосфаты, особенно фосфаты металлов, образующих многозарядные ионы: циркония, титана, хрома, железа, сурьмы, тория, ниобия, гафния и т. п. Синтезированы и смешанно-фосфатные иониты (фталофосфат и сульфосалицилфосфат циркония). Особенно широко изучался цирконилфосфат (ZrO)m
·(Н2PO4)n
, где отношение m :n меняется в зависимости от условий синтеза. Цирконилфосфат устойчив в концентрированных кислотных и солевых растворах, в которых он сохраняет значительную способность к селективной сорбции ряда катионов. Для него характерны термическая и радиационная устойчивость, высокая емкость обмена, высокая селективность к ионам щелочных металлов, меняющаяся в ряду:
Li+
< Na+
< К+
~ NH4-
< Rb+
< Сs+
Высокая селективность к цезию важна при переработке сильнокислых и засоленных отходов атомных реакторов, содержащих долгоживущий изотоп 137Cs. Цирконилфосфат хорошо сортирует ионы таллия, значительно хуже — ионы щелочноземельных металлов из кислых растворов, еще слабее — ионы редкоземельных металлов.
Полимерные комплексные иониты — поли- и гетерополикислоты и их соли — полимолибденовая и поливольфрамовая кислоты, фосформолибдаты и фосфорвольфраматы, ферроцианидмолибдаты и вольфраматы и т. д. — обладают довольно сложной ажурной кристаллической структурой, содержащей каналы и полости. Весьма важны комплексные цианиды, в первую очередь ферроцианиды (железа, меди, цинка, молибдена, вольфрама, никеля, ванадия, урана, титана, олова и т. п.) и смешанные ферроцианиды типа гексацианокобальтоферрита калия. Большой интерес вызывают производные полисурьмяной кислоты (проявляющей свойства сильной кислоты) и производные смешанных фосфорносурьмяной, кремнесурьмяной и титаносурьмяной кислот. Ионообменными свойствами обладают многие нерастворимые соли- сульфиды тяжелых металлов, оксалаты церия, хрома циркония.
Неорганические иониты трудно классифицировать и кратко описать — они еще недостаточно изучены, а их селективные свойства зачастую весьма неожиданны и зависят от способа синтеза ионитов. Скажем, для оксалата циркония в виде геля характерен ряд:
Na+
< K+
~ Rb+
~ Cs+
а в кристаллической форме он селективен к иону калия:
Na+
< Cs+
< Rb+
< К+
Фосфат титана обменивает из щелочных ионов лишь ионы Li+
, Na+
и NH4
+
и не сортирует ионы К+
, Rb+
и Cs+
. Для гидроксида циркония характерен обратный ряд:
Cs+
< Rb+
< K+
< Na+
< Li+
так как она сортирует эти ионы в негидратированной форме. Оксид титана также проявляет избирательность к иону лития, некоторые иониты проявляют селективность к ряду других, самых различных ионов (полисурьмяная кислота — к Ag+, Pb2+, оксалат циркония к ионам Fe3+, Pb2+).
Часто неорганические иониты мелкокристалличны, и это затрудняет их применение. Однако с помощью связывающих веществ они могут быть получены в виде гранул, пропитанных этими ионитами бумаг, волокнистых материалов и даже зерен сорбентов и обычных органических смол.
Большинство синтезированных ионитов используется в аналитической и препаративной химии с целью упростить разделение относительно небольших количеств трудно разделяемых ионов или для получения чистых веществ и препаратов. Кроме того, они представляют существенный познавательный интерес, Многие природные иониты силикатного и алюмосиликатного характера (обычные и модифицированные силикагели, глины, вермикулит, туфы, перлит, выветренные базальты и т. п.) применяются для очистки промышленных стоков и отходов, как искусственные почвы при выращивании растений в теплицах и для ряда других целей.
Все же самая важная, но недостаточно изученная роль природных неорганических ионитов – их участие в геохимических процессах на поверхности земли, на дне океанов, а также в геотремальных и магматических процессах, в том числе при образовании полезных ископаемых [9].
Иониты и ионный обмен
Химические реакции могут быть реакциями присоединения, разложения, обмена, и замещения. Последняя реакций наиболее сложна и обширна. Участвующие в них разные молекулы обмениваются молекулярными группировками и атомами, часто изменяя при этом их химическое состояние и строение.
Химические реакции могут быть гомеофазными, т. е. протекающими в однородной среде. Существует еще множество гетерофазных реакций, в которых реагирующие вещества сначала находятся в разных фазах, и течение реакции связно с переносом веществ между фазами. В более узком обычном смысле под ионным обменом подразумевают обмен образующимися при диссоциации электролитов ионами между фазами гетерогенной системы, по крайней мере одна из которых обладает особыми свойствами и называется ионитом.
Ионит – это вещество или совокупность веществ, обладающая следующими свойствами:
1. Он образует отдельную фазу гетерогенной системы.
2. по крайней мере одно из образующих эту фазу веществ способно диссоциировать на ионы и является электролитом.
3. По крайней мере одна из разновидностей ионов в ионите по различным причинам содержится только в фазе ионита, не может ее покинуть и перейти границу раздела.
Эти ионы называют закрепленными, или фиксированными, ионами, «зарядами», функциональными группами и т.п. Фиксированными ионами могут быть катионы, анионы и вместе – те и другие. Чаще всего фиксированные ионы образуются в ионите при диссоциации включенных в ионит фиксированных ионогенных (катионогенных и анионогенных) групп.
4. Фаза ионита макроскопически электронейтральна и в условиях равновесия не содержит свободного электрического заряда.
Итак, в обычном понимании:
1. Ионный обмен – гетерогенный химический процесс с участием электролитов и образуемых ими ионов. Одной из фаз этой гетерогенной системы обязательно является ионит.
2. Ионит – особая фаза, удерживающая в себе закрепленные ионы, которые не могут переходить ее границу, но способная к обмену с наружной средой другими ионами (противоионами).
С точки зрения термодинамики, фаза ионита частично закрытая система, обменивающаяся с внешней средой лишь некоторыми из своих составляющих (компонентов). Равновесие ионита с внешней средой — неполное равновесие, устанавливаемое лишь относительно тех составляющих системы, которые переходят межфазовую границу.
В зависимости от того, какие ионы способны обменивать иониты с внешней средой, последние можно разделить на следующие группы:
1) катиониты — иониты с закрепленными анионами и анионогенными группами, обменивающиеся с внешней средой катионами;
2) аниониты — иониты, содержание закрепленные катионы или катионогенные группы, обменивающиеся анионами;
3) амфолиты — иониты, содержащие закрепленные катионогенные и анионогеиные группы и в определенных условиях выступающие либо как катиониты, либо как аниониты.
B более широком понимании, ионным обменом являются любые процессы обмена (перераспределения) ионов между электролитами с участием достаточно сложных молекул электролитов (в первую очередь, полиэлектролитов), а также образуемых ими структурных объединений, агрегатов и коллоидных частиц.
Ионообменными являются многие гомеофазные процессы, а также процессы с участием коллоидных образований, которые могут рассматриваться и кик гетерофазные (с учетом роли их поверхности) и как гомеофазные, Часто отнести такие системы к гетеро или гомеофазным трудно еще и потому, что сам ионный обмен существенно влияет на образование или распад того или иного структурного объединения частиц. В этом случае важны поверхностные явления, которыми в теории гетерофазного ионного обмена пренебрегают.
С ионным обменом связаны многие процессы с участием мыл, поверхностно – активных веществ, красителей, биополимеров – белков, ферментов, нуклеиновых кислот, нуклеопротеидов, липидов, с функционированием разнообразных биологических структур и мембран. Важность ионного обмена в этих системах неоспорима, хотя и не всегда осознана и изучена.
Строго говоря, к ионному обмену относится и обмен ионами между простыми электролитами, но по традиции его рассматривает теория растворов электролитов и химия комплексных соединений [10].
Обмен на анионитах.
Анионный обмен лучше всего изучен на сильно основных анионитах с матрицей из полистирола и сшивкой из дивинилбензола с группой R-[N(CH3)3]+
OH-
-сильным органическим основанием. Как и сульфополистирольные катиониты, эти часто применяемые на практике иониты — простейший и наименее специфический объект. Хотя им посвящено немало теоретических исследований, однако понимаете механизма обмена еще не вполне достигнуто. Для более простого случая обмена однозарядных галоген-анионов установлен ряд селективности: F-
< Cl-
< Вr-
< I-
, аналогичный ряду для щелочных металлов на катионите. Однако, для высших членов ряда наблюдаются большие коэффициенты селективности. По-видимому, это связано с изменением гидратации анионов при их взаимодействии с функциональной группой в ионите. Так же, как и для катионного обмена, увеличение степени сшивки, уменьшающее набухание, повышает селективность обмена.
В литературе приводятся ряды селективности для разных анионов. Вот один из таких рядов, согласующийся с рядом для галоген ионов:
OH-
<F-
< NH2
CH2
COO-
< CH3
COO-
< HCOO-
< Н2
РО4
-
< HCO3
-
<Cl-
<NO2
-
<HSO3
-
<CN-
<Br-
<NO3
-
< НSО4
-
< I-
< цитрат-ион < салицилат-ион
Обращает на себя внимание низкая селективность сильноосновных анионитов к ОН-
-иону, означающая, что их формы — сильные основания.
Данные о сорбции анионов на слабоосновных анионитах едва ли поддаются обобщению. Аниониты нашли широкое применение в пищевой, свеклосахарной, фармацевтической промышленности и в металлургии редких металлов. Многие металлы сорбируются анионитами в виде образуемых ими в растворах анионных комплексов. Это используется в металлургии цветных и редких металлов (цианидные комплексы золота и серебра, комплексы платины и т.п.).
Роль комплексообразования важна и при сорбции примесей Cu2+
,Co2+
, Zn2+
, Сг3+
и Pb2+
анионитами из хлоридных растворов. При повышении концентрации хлоридов в растворе образуются хлоридные комплексы, неустойчивые в разбавленных растворах, и сорбция этих металлов сильно увеличивается.
Некоторые аниониты содержат функциональные группы, способные к непосредственному комплексообразованию с катионами ряда металлов. Таков механизм селективной сорбции ионов меди и других переходных металлов из щелочных (аммиачных) растворов анионитом ЭДЭ-10П, используемым для удаления меди из сточных вод [13].
1.3.
Селективные и комплексообразующие иониты.
Идея синтеза ионитов, селективных к определенным ионам или группам ионов, возникла давно. С этой целью пытались вводить в ионит в процессе синтеза вещества, способные к комплексообразованию, присоединять к матрице полимера в качестве функциональных групп специфические реактивы на определенные ионы: диметилглиоксим, дипикриламин, меркаптаны, N-метилглюкамид и т. п. Однако часто их селективные свойства значительно ухудшаются при введении в матрицу.
Другое направление синтеза комплексообразующих ионитов — включение в матрицу комплексонов, способных к образованию циклических внутрикомплексных соединений (хелатов) и прежде всего иминодиуксусной кислоты и ее производных. Таковы иониты с присоединенными группами оксихинолина, иминодиуксусной, этилендиаминтетрауксусной, этнлендиаминдипропионовой, бензилиминодиуксусной, саркозиновой и аминоэтилфосфоновой кислот.
Важное свойство этих ионитов – возможность управления их селективностью путем изменения внешних условий, в частности рН, поскольку. Как правило, селективные функциональные группы обладают свойствами кислот или оснований.
Многие комплексообразующие смолы имеют плохие кинетические свойства из-за малой диссоциации групп, а следовательно, и плохого набухания.
Чем селективнее сорбируется ион, тем труднее извлечь его из ионита. Тем больше время и затраты реактивов необходимы на десорбцию. В связи с этим важны работы по модификации комплексообразующих ионитов – синтезу ионитов с развитой поверхностью (пористых), ионитов, содержащих кроме специфических функциональных групп полярные группы, либо неспецифические, но легкие гидратируемые функциональные группы (например, сульфогруппы). На комплексообразующих ионитах значительно существеннее явление сверхэквивалентного обмена вследствие слабой диссоциации комплексов.
При сорбции ионов комплексообразующими ионитами функциональные группы часто далеко не полностью насыщают координационные связи иона-комплексообразователя. Оставшиеся координационный связи насыщаются за счет дополнительного связывания лигандов молекул или ионов из внешнего раствора.
Если в растворе присутствуют разные лиганды, то они конкурируют друг с другом за место в координационной сфере сортированного ионитом иона-комплексообразователя, обмениваются друг с другом (лиганд-обмен). В качестве катионов-комплексообразователей обычно применяются Ag+
, Cu2+
, Zn2+
, Ni2+
, Cd2+
, Fe3+
и др. Например, сорбированные ионитом ионы меди в аммиачной среде могут обменивать молекулы аммиака на пиридин, тиомочевину, 1,3-диаминопропенол и т.д. Если лиганд — ион, то при лиганд-отмене катионит работает как анионит (и наоборот). В случае нейтральных лигандов ионит работает как молекулярный сорбент при полном его насыщении в процессе конкурентной сорбции молекул.
Законы лиганд-обмена сходны с ионным обменом. Так, при обмене лигандов, занимающих разное число мест в координационной сфере, наблюдается концентрационно-валентный эффект — увеличение сорбции полидентатного лиганда при разбавлении внешнего раствора.
Лиганд обмен возможен не только на комплексообразующих, но и на обычных ионитах (в том числе на сульфокатионитах), насыщенных комплексными ионами. Однако здесь гораздо существеннее роль ионного состава внешнего раствора, изменение которого приводит к десорбции этих ионов. При их специфической сорбции ионитом-комплексообразователем это явление менее существенно. Поэтому лиганд-обмену на комплексообразующих смолах не препятствует значительная концентрация солей во внешнем растворе.
Основанная на лиганд-обмене лиганд-хроматография – тонкий метод, пригодный для весьма сложных разделений, например для разделение аминов, аминокислот, пептидов, производных нуклеиновых кислот, эфиров непредельных кислот, амфетаминов и дикетонов [15] .
В настоящее время большое внимание уделяется повышению селективности ионообменных процессов за счет использования селективных ионообменников. Последние получают введением соответствующих функциональных групп (производных органических реагентов) в полимерную матрицу. Функциональные группы этих ионообменников обладают способностью образовывать комплексы или хелаты с некоторыми ионами и благодаря этому (при соответствующей обработке анализируемого раствора) селективно поглощают один вид или ограниченную группу ионов из сложных смесей ионов.
Например, смолы, содержащие группы – N(CH2
COOH)2
, селективно поглощают следовые количества тяжелых металлов в присутствии больших количеств ионов щелочно-земельных металлов. Ионообменник, содержащий группы – РО(ОН)2
, в кислой среде селективно поглощает скандий – редкоземельные элементы.
Другую группу ионообменников, проявляющих повышенную селективность к некоторым ионам, образуют неорганические сорбенты. Недостатками этих ионообменников являются низкая устойчивость большинства из них в щелочных растворах, склонность к пептизации, малая обменная емкость, а также трудности, связанные с получением их в форме, удобной для работы в динамическом режиме [16].
Жидкие иониты
Одним из наиболее совершенных методов разделения вещества является экстракция — избирательное извлечение веществ из смесь при распределении растворенных веществ между двумя несмешивающимися друг с другом растворителями (как правило, между водой и органическим растворителем). Экстрагентами часто являются неполярные органические растворители. Обычно они слабо извлекают из внешнего раствора ионизированные соединения. Однако извлечение ионов можно резко усилить, растворив вещество с молекулами двоякой — гидрофобно-гидрофильной (дифильной) — природы. Они обладают способной к диссоциации функциональной группой (гидрофильной головкой) и одним или несколькими гидрофобными неполярными хвостами, достаточно длинными для того, чтобы «затащить» полярную группу в неполярный растворитель. Из неполярного растворителя эти вещества в водную фазу практически не переходят. В органической фазе они в некоторой степени сохраняют способность к диссоциации и могут обмениваться ионами с внешним водным раствором. Диссоциация подобных растворенных веществ в органическом растворителе существенно усиливается при контакте с водой, так как полярная группа собирает вокруг себя молекулы воды, в некотором количестве проникающие в органической растворитель, «затаскивая» воду в растворитель.
Такие смешанные экстрагенты называют жидкими ионитами, так как происходящие при экстракции ими ионов явления весьма сходны с ионным обменом на твердых ионитах.
Среди жидких ионитов также есть катиониты и аниониты. Анионитами, например, являются растворы четвертичных алкиламмониевых оснований и их солей (соли тетраоктиламмония, метилтрилауриламмония и т.д.),длинноцепных аминов (триоктиламина, трилаурнламина). Катиониты — растворы фосфорорганических кислот 1ди-2-этилгексилфосфорпая кислота), жирных кислот и их солей, алкилсульфокислот (динонилнафталинсульфокислота) и т. п.
Жидкие иониты расширяют пределы экстракционного метода, вовлекая в него ионообменные процессы, а возможности метода ионного обмена увеличиваются благодаря новым ионитам с иными физическими, термодинамическими и кинетическими свойствами.
Жидкие иониты особо интересны для теории ионного обмена. В самом деле, здесь отсутствуют такие влияющие на ионный обмен факторы, как сшивка полимерных цепей, да и сами цепи значительно короче. В отличие от твердых, жидкие иониты химически и физически однородны. Появляется возможность свободно изменять в широких пределах емкость обмена, диэлектрическую постоянную и другие свойства, влияющие на селективность ионного обмена.
Жидкие иониты значительно проще исследовать физическими и физико-химическими методами; легко, например, измерить поверхностное натяжение (в том числе и поверхности раздела двух растворителей), электродные потенциалы и активности ионов.
Следует помнить, что возможны и новые эффекты, а эффекты, наблюдающиеся у обычных ионитов, могут проявляться в существенно иной степени.
Жидкие иониты способны к обмену обычных и комплексных анионов. Так, для 0,1н. раствора темраоктиламмонийбромида (соль сильного основания) в толуоле установлен ряд:
OH-
<F-
<СН3
СОО-
<НСО3
-
<НSО4
-
<Сl-
<
< Вr-
< С6
Н5
СОО-
< NО3
-
< I-
< СlO4
-
Как и для анионитов, здесь можно предположить существенное влияние различной гидратации ионов в обеих фазах на селективность обмена. Растворы солей четвертичных аммониевых оснований в керосине характеризуются следующим рядом селективности для комплексов ионов-комплексообразователей:
Ni2+
< Mn2+
< Со2+
< Сu2+
< РЬ2+
< Zn2+
< Cd2+
Недостаточно ясно, отражает ли этот ряд только собственно ионообменные явления, либо на него влияет и неполное связывание ионов в комплексы. Известно, например, что хлоридные комплексы металлов легко диссоциируют по реакции типа:
[ZnCl4
]2-
= Zn2+
+ 4Cl-
при концентрации Cl-
в растворе < 0,4н и, следовательно, уже не участвуют в анионном обмене.
Жидкие аниониты способны к экстракции кислот из водных растворов. Этот процесс можно представить и как ионный обмен. Например, сорбцию кислоты триоктиламином можно записать как в виде
R3
N + Н+
+ Сl-
= R3
NHCl
так и в виде:
R3
N · HOH+Н+
+ Cl-
= R3
N · HCl + Н2
О
Исследование жидких ионитов методами ионного обмена, несомненно, должно привести к важным теоретическим и практическим результатам.
Жидкие иониты широко применяются в металлургии благородных металлов, кобальта, никеля, меди при очистке сточных вод, переработке продуктов деления урана и плутония и в других радиохимических процессах. Как правило, использование, особенно в крупнотоннажном производстве, даже весьма экзотических экстрагентов, при усовершенствовании технологии их синтеза оказывается экономически выгодным.
Жидкие иониты применяют в аналитической химии для разделения ионов и в качестве мембран ионселективных электродов. В перспективе возможно их применение в виде селективных мембран для разделения ионов методом мембранной технологии [17].
1.4. Основные характеристки ионообменников.
а) Сильнокислотные катионообменники.
Выпускают два типа ионообменников, содержащих группы — SO3
H, на стирол-дивинилбензольной и фенолформальдегидной матрицах. Группы SO3
H связаны с бензольным ядром непосредственно или через метиленовую группу. По степени ионизации ионогенных групп ионообменники сравнимы с сильными минеральными кислотами. Группы — SO3
H, связанные непосредственно с бензольным кольцом, диссоциируют легче, чем группы — СН2
SO3
Н.
При контакте ионообменников в Н-форме с растворами нейтральных полей ионы Н+ переходят в раствор, а ионообмениики превращаются в соответствующие солевые формы. Химические реакции ионообменников подобны реакциям серной кислоты. Обменная емкость смол практически не зависит от рН раствора, ионообменники могут быть использованы в кислых, нейтральных и щелочных растворах.
Селективность групп — SO3
H повышается с увеличением атомного номера, валентности и степени ионизации обмениваемых ионов и понижается с увеличением ионного радиуса гидратированных ионов. Как правило, селективность уменьшается в рядах (Dowex 50-X8):
Ag+
> Cs+
> Rb+
> К+
> NH+
> Н+
> Li+
;
Ba2+
> Sr2+
> Сa2+
> Mg2+
> Be2+
;
Ва2+
>РЬ2+
>Sr2+
>Са2+
>Ni2+
=Cu2+
>Cd2+
>Co2+
>Zn2+
=Mg2+
>Mn2+
>Be2+
>UO2
2-
> Hg2+
; La3+
> Се3+
> Cr3+
;
Th4+
(NO3-
)4
>Fe3+
>Al3+
>Ва2+
>Tl+
(SO4
2-
)=Pb2+
>Sr2+
>Са2+
>СО3
2-
>Ni2+
=Cu2+
>Zn2+
= Mg2+
>UO2
2-
(NO3
-
)2
=Mn2+
>Ag+
>Cs+
>Ве2+
(SO4
2-
)=Rb+
>Cd2+
>NH4
+
=К+
>Na+
>Н+
>Li+
Эти ряды селективности справедливы для разбавленных растворов (приблизительно 0,1 моль/л хлоридов, если не указаны другие условия). В концентрированных растворах однозарядные ионы поглощаются лучше, чем многозарядные.
б) Среднекислотные катионообменники.
В настоящее время выпускают ионообменники, содержащие группы — РО(ОН)2
или – ОРО(ОН)2
, на различных полимерных матрицах. Химические свойства ионообменников подобны свойствам фосфористой или фосфорной кислот. По способности к диссоциации ионогенных групп смолы в Н-форме занимают промежуточное положение между сильнокислотными и слабокислотными катионообменниками.
Обменная емкость ионообменников зависит от рН внешнего раствора. Большая часть одно- и двухзарядных катионов наиболее эффективно поглощается при рН > 5. На селективность ионогенных групп большое влияние оказывает природа сорбируемого иона и рН раствора:
—РО(ОН)2
: Pb2+
> Cu2+
> Zn2+
> Cd2+
> Mn2+
> Со2+
> Ni2+
;
диаллилфосфат: Н+
> Ag+
> Cs+
> Rb+
> K+
> Na+
> Li+
(в кислой среде);
— РО(ОН)2
: Cs+
> RЬ+
> К+
> Na+
> Li+
(pH 6,7 — 8,5)
Cs+
> Rb+
> К+
> Li+
> Na+
(рН 10,0);
Li+
> Na+
≥ RЬ+
≥ Cs+
> К+
(рН 12,6);
— РО(ОН)2
:Th4+
> U4+
> UO2
2-
> Fe3+
> РЗЭ > Н+
> Cu2+
> Cu2+
> Cd2+
> Mn2+
>
Co2+
> Ni2+
> Са2+
> Mg2+
> Sr2+
> Ва2+
> Na+
Для полного превращения ионообменников в Н-форму из форм тех ионов, которые располагаются в ряду селективности после ионов Н+
, необходим небольшой (по сравнению со стехиометрией) избыток сильной минеральной кислотой. Для других ионных форм требуется значительно больший избыток кислоты. Второй путь превращения смолы в Н-форму — предварительное элюирование катионов подходящим комплексообразующим веществом.
в) Слабокислотные катионообменники.
Выпускают монофункциональные ионообменники, содержание группы -CОOH, на основе сополимеров акриловой и метакриловой кислот с дивинилбензолом и ионообменники с группам -СООН и -ОН, полученные поликонденсацией фенолов с резорциловой кислотой. Ионообменники в Н-форме не выделяют ионы Н+
при контакте с растворами нейтральных солей; по степени ионизации соответствуют уксусной кислоте.
Обменная емкость ионообменников сильно зависит от рН раствора. Наиболее эффективная область рН находится в пределах рН 6 — 14.
Характерным свойством ионообменников являются высокая селективность к ионам H+
и относительно высокое сродство к ионам щелочноземельных металлов. Ряды селективности для ионов металлов имеют обратный порядок по сравнению с сильнокислотными ионообменниками.
Ряд селективности при рН 7: Mg2+
< Са2+
< Ni2+
< Со2+
< Cu2+
;
г) Сильноосновные анионообменники.
Выпускают ионообменники, содержащие функциональные группы – N+
(СН3
)3
Сl (тип I), — N+
(СН3
)2
С2
Н4
ОН·Cl (тип II) или пиридиновые группы, преимущественно на основе сополимеров стирол-ДВБ. Ионообменники в ОН-форме вытесняют из растворов нейтральных солей анионы; в результате этого обмена ионообменники превращаются в соответствующие солевые формы, а в растворе образуются гидроксиды. По степени ионизации ионогенных групп ионообменники сравнимы с гидроксидами щелочных металлов. Ионообменники в ОН-форме поглощают даже слабодиссоциированные кислоты (борную, кремневую).
д) Среднеосновные анионообменники.
Ионообменники содержат как сильноосновные, так и слабоосновные ионогенные группы (преимущественно группы третичных аминов). Регенерированные раствором гидроксида натрия, ионообменники вытесняют анионы из растворов нейтральных солей и сорбируют слабые кислоты пропорционально содержанию сильноосновных групп. После регенерации растворами карбоната натрия или гидроксида аммония ионообменники ведут себя как слабоосновные.
ж) Слабоосновные анионообменники.
Полимерной основой ионообменников, содержащих ионогенные группы – NH3
, – NHR, – NR1
R2
(первичные или вторичные амины), являются стирол-дивинилбензольные, полиамин-эпихлоргидринные и фенолформальдегидные матрицы. По степени ионизации ионогенных групп ионообменники сравнимы с гидроксидом аммония.
Аминогруппы ионообменников склонны к образованию прочных комплексов с Ag+
, Cu+
и другими катионами.
Сродство анионов к ионообменникам уменьшается в ряду ОН-
> SO4
2-
> СгO4
2-
>цитрат >тартрат >NO3
-
>AsO4
2-
>РО4
3-
>МoO4
2-
>СН3
СОО-
>I-
=Вг-
>Cl-
>F-
[16].
1.5. Ионообменные процессы
Для того чтобы начался ионообменный процесс, необходимо ионообменную смолу привести в контакт с раствором, содержащим способные к обмену ионы. Существуют два метода осуществления контакта ионообменника ионами в растворе: статический (встряхивание) и динамический (колоночный).
Статический метод
: Ионообменник перемешивают или встряхивают с раствором в подходящем сосуде. После достижения равновесия между ионообменником и ионами и растворе фазы разделяют фильтрованием, декантацией или центрифугированием и анализируют на содержание в них ионов. Количественное поглощение ионов из раствора может быть достигнуто при большом избытке ионообменной смолы (одностадийный статический процесс) или последовательным прибавлением небольших количеств смолы в раствор. После установления равновесия каждая порция смолы отделяется от раствора. Этот метод, называемый многостадийным (каскадным) статическим процессом, является лабораторным, его применение ограничено вследствие большой затраты времени и возможности экспериментальных ошибок.
Многостадийный процесс целесообразно использовать при анализе систем, в которых в результате ионного обмена выделяются газы, а также в тех особых случаях, когда необходимо сдвинуть равновесие в сторону ионообменного процесса. Примерами таких процессов служат реакции нейтрализации, образования устойчивых комплексов и нерастворимых соединены, превращения нерастворимых веществ в растворимые формы. Например, барий может быть переведен в раствор из нерастворимого сульфата
бария
BaSO4
+ 2RSO3
Na = (RSO3
)2
Ba + Na2
SO4
Смолу промывают водой и элюируют ионы бария 3 — 4М соляной кислотой. Процесс проводят в присутствии большого избытка смолы при повышенной температуре. Аналогичную методику используют при анализе сульфатов свинца, стронция или кальция, хлорида свинца, нерастворимых фосфатов двухвалентных металлов и т. п.
Одностадийный статический процесс иногда применяют в качественном анализе: ионы концентрируют на белой или слабоокрашенной смоле и выполняют цветную реакцию на соответствующий ион непосредственно в фазе смолы.
Одной из наиболее важных областей применения статического метода является определение различных физико-химических параметров: структуры, устойчивости комплексов, коэффициентов селективности, равновесных характеристик для динамических опытов.
Динамический метод.
Ионообменную смолу (набухшую в воде или подходящем растворителе) в виде гомогенной смеси с раствором помешают а вертикальную колонку и пропускают анализируемый раствор. Другие методы пропускания раствора через колонку (противоточный) редко применяют в аналитической практике. Проведение ионообменных процессов в колонках обеспечивает возможность количественного обмена ионов из раствора и разделение смесей с максимальной эффективностью.
Общая схема ионообменного процесса в колонках включает стадии сорбции, промывание соответствующим растворителем или раствором (чаше всего водой), регенерацию (элюирование сорбированных ионов). Представляют интерес особые случаи применения этих операций в аналитической химии.
а) Селективная сорбция. Этот метод основан на выборе подходящих условий сорбции для одного элемента или для небольшой группы элементов, присутствующих в смеси. Для удержания нежелательных компонентов смеси в растворе часто применяют вещества, которые образуют с мешающими элементами достаточно прочные несорбируемые комплексы.
Например, для превращения кадмия (в смеси Zn - Cd) и железа в несорбируемые сильнокислотными катионообменниками комплексы вводят иодиды и цианиды соответственно; часто в качестве комплексообразующих веществ используют такие комплексоны, как этилендиаминтетрауксусная кислота (ЭДТА), этиленгликольтетрауксусная кислота (ЭГТА) и др.
Метод селективной сорбции применяется также при разделении элементов на анионообменниках. Например, из растворов соляной кислоты, содержащей смесь элементов, при соответствующей концентрации кислоты на смоле может удерживаться только один ион в то время как другие ионы проходят через колонку (отделение Fe от Al, Co от Ni и т.п.).
б) Селективное элюирование. При селективном элюировании наблюдается картина, обратная селективной сорбции. Задача заключается в выборе условий, при которых один тип ионов десорбируется, а другие прочно удерживаются смолой. Процесс разделения ускоряется при использовании коротких колонок.
Процесс разделения в методе селективного элюирования основаны, как правило, на различиях в константах устойчивости разделяемых ионов. Типичным примером успешного применения метода селективного элюирования является разделения смеси Ni – Mn – Co – Cu – Fe – Zn элюированием соляной кислотой (с анионообмеников).
Сочетание селективной сорбции и селективного элиюрования значительно упрощает процесс разделения сложных смесей [18].
Хроматографический метод.
Разделение сложных смесей на индивидуальные компоненты возможно только при наличии высокоэффективных методов. Хроматографические колоночные методы подразделяются на три основных вида: фронтальный вытеснительный и элюентный анализ. Фронтальный и вытеснительный виды хроматографического анализа имеют ограниченное значение при разделении смесей неорганических ионов, так как не позволяют количественно разделить смеси на индивидуальные компоненты.
Метод элюентной хроматографии основан на поглощении анализируемой смеси ионов в верхней части колонки в виде тонкого слоя и разделения с помощью соответствующего элюирующего раствора при продвижении его по колонке сверху вниз. В процессе перемешивания раствора состав поглощенной пробы непрерывно изменяется: ионы, имеющие более низкое сродство к ионообменнику – медленнее. После пропускания достаточного количества элюента индивидуальные компоненты анализируемой смеси распределяются вдоль ионообменной колонки в виде отдельных зон.
В ионообменных процессах могут быть использовать не только гранульные ионообменники, но также материалы в форме бумаги, тонких пластин или мембран. Ионообменную бумагу получают введением тонкодисперсных частиц смолы в бумажную пульпу или непосредственно в слое бумаги.
Практические методы работы с ионообменными материалами в форме бумаги, тонких пластин и мембран аналогичны приемам, используемым в бумажной и тонкослойной хроматографии и в электрохимических методах разделения [20].
2.
Экспериментальная часть.
2.1. Характеристика исходных веществ. Методика химических и физико-химических методов исследования.
В работе были использованы в качестве исходного сырья:
1. CoСО3
2.К4
P2
O7
3. Fe2
O3
4. NH4
Н2
PO4
5. MnSO4
· 5H2
O
6. NiSO4
·6H2
O
Все вещества были взяты марки «ч.д.а.»
Для проведения химического анаиза применялись следующие реактивы [6]:
1) Азотная кислота, HNO3
, конц., разбавленная 1:2;
2) Метаванадат аммония, NH4
VO3
, 0,25%-ный раствор;
3) Молибдат аммония, (NH4
)6
Mo7
O24
· 4H2
O, 5%-ный раствор;
4) Реактив на фосфаты;
5) Стандартный раствор дигидрофосфата калия, реактив А (KH2
PO4
);
6) Лимонная кислота, 2%.
Все вещества были взяты марки «ч.д.а.»
Для определения пентоксида фосфора применялись методика, принятая при анализе удобрений [6].
Химический состав катионов (содержание Ni2+
, Со2+
, Fe2+
) определяли методом атомно-адсорбционного анализа.
Фазовый состав синтезированных соединений определялся снятием рентгенограмм на дифрактометре ДРОН-05 с использованем медного излучателя.
ИК спектры поглощения записывали в диапозоне 4000 – 500 см-1
с помощью спектрофотометра Sресоrd, используя для съемки образцы (0,5 масс.%) в виде таблеток КВr.
Фотоколориметрический метод определения Р2
О5
.
Реактивы: серная кислота, отн.пл. 1,84 г/см3
; соляная кислота - 20%-ный раствор; азотная кислота, отн.пл. 1,4 г/см3
и разбавленная 1:2.
Метаванадат аммония (0,25%-ный раствор) – взвешивают 2,5г NH4
VO3
с точностью до 0,01г, растворяют в 500мл горячей воды, приливают 20 мл азотной кислоты, отн.пл. 1,4 г/см3
, доводят объем до 1л и фильтруют.
Молибдат аммония (5 %-ный раствор) – взвешивают 50г (NH4
)6
Mo7
O24
· 4H2
O с точностью до 0,01г, растворяют в 500 мл воды при 500
С; после охлаждения доводят объем до 1л и фильтруют.
Реактив на фосфаты: смешивают равные объемы растворов азотной
кислоты 1:2, метаванадата аммония и молибдата аммония;
Стандартный раствор дигидрофосфата калия, реактив А (растворяют 1,9175г КН2
РО4
в дистилированной воде в мерной колбе на 1л , прибавляют 10 мл серной кислоты отн.пл. 1,84 г/см3
, доводят объем водой до метки и перемешивают; в 1 мл раствора А содержится 1 мг Р2
О5
).
Построение калибровочного графика.
В шесть мерных колб вместимостью 100мл вводят микробюреткой 1, 2, 3, 4, 5, 6 мл раствора дигидрофосфата калия, что соответствует 1, 2, 3, 4, 5, 6 мг Р2
О5
. Объем раствора в каждой колбе доводят до 20 мл водой, добавляют 25мл реактива на фосфаты (А), разбавляют до метки водой и через 5 мин. Измеряют оптическую плотность относительно раствора сравнения, содержащего 1 мг Р2
О5
. Калибровочный график периодически проверяют по трем точкам, например 1,5; 1,75; 2,0 мг Р2
О5
. Измерения на калориметре следует проводить при температуре воздуха от 10 до 350
С.
Рабочие поверхности кювет перед каждым измерением необходимо тщательно протирать спирто-эфирной смесью. Жидкость в кюветы наливают до метки на боковой поверности кюветы. При установке кювет в кюветодержатели нельзя касаться пальцами участков поверхностей ниже уровня жидкости в кювете.
Колориметр включают в сеть за 15 мин до начала измерений. Во время прогрева кюветное отделение должно быть открыто (при этом шторка перед фотоприемником и перекрывает световой пучок). В световой пучок помещают кювету со стандартным раствором, по отношению к которому производятся измерения, и крышку кюветного отделения закрывают. Измерения проводят 3-5 раз и окончательное значение измереной величины определяют как среднее арифматическое из полученных значений.
Ход анализа.
В мерную колбу вместимостью 100 мл отбирают объем фильтрата (фильтрат получают также как и при определения Р2
О5
весовым методом), соответствующий 0,7-1,75 мг Р2
О5
и разбавляют водой до 20 мл. Затем добавляют 25 мл реактива на фосфаты, разбавляют до метки водой. Далее анализ продолжают, как при определения калибровочной кривой.
Расчет.
Содержание Р2
О5
– в массовых долях в % (ω Р2
О5
) вычисляют по формуле:
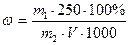 , где , где
m1
– масса Р2
О5
, найденная по калибровочному графику, мг;
m2
– масса анализируемого вещества, г;
V – объем растворы, взятой на анализу, мл.
2.2. Синтез дифосфатов Со,
Ni
и
Fe
и их характеристика.
Дифосфат кобальта
светло-фиолетового цвета получен в системе СоО – Р2
О5
при нагревании смеси СоО (СоСО3
) и NН4
Н2
PO4
до 10000
и выше при соотношении СоО : Р2
О5
= 2:1.
2СоСО3
+ 2NН4
Н2
PO4
→ Со2
Р2
О7
+ + 2NН3
+ 2СО2
+ 3Н2
O
Для Со2
Р2
О7
известны низко- и высокотемпературная модификации, взаимопереход между которыми, осуществляется при 295-3000
С. Теплота модификационного перехода оценивается величиной (7,1±0,8) кДж/моль [22, 39].
Структура низкотемпературной формы α - Со2
Р2
О7
показывает, что угол между связами Р – О – Р = 142,60
[39].Несимметричное строение группы Р2
О7
связывается с различным окружением тетраэдров РО4
катионами: первый тетраэдр окружен пятью ионами кобальта, второй – шестью. Высокотемпературная форма β - Со2
Р2
О7
(как и дифосфаты Ni, Mn, Zn, Mg) изоморфна минералу тортвейтиту. Группа Р – О – Р в этой форме дифосфата кобальта имеет линейное строение, причем а0
и с0
вдвое меньше по сравнению с низкотемпературной формой [40].
Фосфаты железа
представляют интерес в качестве возможных компонентов высокотемпературных фосфатных связок и бетонов. Известно, что мета-, пиро- и ортофосфаты щелочных, щелочноземельных и некоторых переходных металлов при высоких температурах диссоциируют с отщеплением Р2
О5
. В связи с изложенным представляло интерес исследовать поведение FePO4
при нагревании [27].
FePO4
при высоких температурах частично диссоциирует по уравнению:
2FePO4
= Fe2
P2
O7
+ 0,5О2
.
Следовательно, в отличие от фосфатов, щелочных, щелочноземельных и других переходных металлов, которые диссоциируют с отщеплением Р2
О5
, средний ортофосфат железа (III) при нагревании до 1100 – 12000
частично диссоциирует с отщеплением кислорода и превращается при этом в пирофосфат Fe2
P2
O7
[31, 36].
Дифосфат никеля
синтезирован при смешивании эквимолярных растворов Na4
P2
O7
и NiSO4
. Осадок очищали перекристаллизацией из водного раствора насыщенного SO2
[14]. Дифосфат никеля относится к типу тортвейтита и изоструктурен Mg2
Р2
О7
. При синтезе соединений в системе NiO и Р2
О5
и соотношении Р/Ni = 0,8 – 1,2. Также установлены эти формы дифосфата с различными показателями преломления [26].
2NiSO4
+ Na4
P2
O7
→ Ni2
P2
O7
+ 2Na2
SO4
2
.
3. Идентификация синтезированных фосфатов Со,
Ni
и
Fe
и определение их свойств.
Синтезированы дифосфаты Со, Ni и Feв диапазоне температур 25-11000
С и в соотношениях соединений М2+
(оксиды, хлориды и сульфаты) к реагенту (NН4
Н2
PO4
, К4
P2
O7
, Nа4
P2
O7
) 1:1, 1:2.
Известно, что молярное соотношение 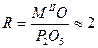 , соответствует дифосфатам, а у циклических фосфатах R = 1. , соответствует дифосфатам, а у циклических фосфатах R = 1.
Согласно, их данным в составе дифосфатов цинка, кобальта, железа и никеля, а также в циклотетрафосфате железа присутствуют катионы Co2+
Ni2+
, Fe2+
соотношение этих катионов которых изменяется в зависимости от условий синтеза.
Таблица 2
Химический состав синтезированных фосфатов
Co
2+
,
Ni
2+
,
Fe
2+
.
| Состав смеси |
Содержание Р2
О5
, % |
Содержание МеО, % |
| эксперимен. |
теоретич. |
эксперимен. |
теоретич. |
| Co2
Р2
О7
|
36,5 % |
48,6 % |
32,0 % |
51,3 % |
| Fe2
P2
O7
|
48,2 % |
49,6 % |
35,0 % |
50,3 % |
| Ni2
P2
O7
|
39,4 % |
49,0 % |
42,0 % |
51,0 % |
Вычисленные значения R для дифосфатов Co, Ni и Fe близко к 2 (RZn2Р2О7
= 2,3
, RCo2Р2О7
= 2,18
, RFe2P2O7
= 2,3
, RNi2P2O7
= 2), для тетрациклофосфата Fe3+
R равна 0,9
.
Идентификацию синтезированных фосфатов осуществляли с помощью методов рентгенофазового анализа, ИК – спектроскопии и химическими методами исследования.
Основной задачей рентгенофазового анализа (РФА) является идентификация различных фаз в их смеси на основе анализа дифракционной картины, даваемой исследуемым образцом. Определение вещества в смеси проводится по набору его межплоскостных расстояний и относительным интенсивностям соответствующих линий на рентгенограмме.
Когерентно рассеянные рентгеновские лучи интерферируют между собой, при этом дифракционной решеткой для рентгеновского излучения служит кристаллическая решетка, поскольку межплоскостные расстояния в кристалле сравнимы с длиной волны излучения.
Целью рентгенофазового анализа является идентификация вещества в смеси по набору его межплоскостных расстояний (d
) и относительным интенсивностям (I) соответствующих линий на рентгенограмме. Для этого, согласно закону Брегга — Вульфа, необходимо определение углов отражения θ [29].
Индивидуальность дифосфатов кобальта, никеля и железа (II) установлена результатами рентгенофазового анализа, которые показали наличие единственной кристаллической фазы, свободной от примесей других фаз (табл. 3; рис1, 2, 3).
Таблица 3
Результаты рентгенофазового анализа для дифосфатов Со2+
,
Ni
2+
и
Fe
2+
,
d · 10-1
, нм
| Co2
Р2
О7
|
| литературные данные |
синтезированный |
| d · 10-1
, нм |
І/І0
, % |
d · 10-1
, нм |
І/І0
, % |
| 3,01 |
100,00 |
3,01 |
100,00 |
| 2,96 |
56,00 |
2,97 |
56,30 |
| 2,55 |
22,00 |
2,54 |
21,90 |
| 2,11 |
24,00 |
2,11 |
24,00 |
| 2,02 |
9,00 |
2,02 |
8,97 |
| 1,60 |
3,00 |
1,61 |
3,02 |
| 1,58 |
16,00 |
1,57 |
15,90 |
| Fe2
P2
O7
|
| 5,63 |
20,00 |
5,45 |
19,37 |
| 4,68 |
30,00 |
4,66 |
29,91 |
| 3,81 |
5,00 |
3,86 |
5,06 |
| 3,43 |
4,00 |
3,42 |
3,99 |
| 3,07 |
70,00 |
3,07 |
70,00 |
| 2,72 |
30,00 |
2,72 |
29,97 |
| 2,02 |
10,00 |
2,02 |
10,01 |
| Ni2
P2
O7
|
| 9,80 |
30,00 |
9,82 |
30,06 |
| 5,52 |
10,00 |
5,50 |
9,96 |
| 3,16 |
100,00 |
3,15 |
100,00 |
| 2,76 |
8,00 |
2,76 |
7,90 |
| 2,54 |
2,00 |
2,54 |
2,00 |
| 1,83 |
10,00 |
1,83 |
10,03 |

Рисунок 1. Рентгенноргамма дифосфат кобальта Co2
Р2
О7

Рисунок 2. Рентгенноргамма дифосфат железа Fe2
P2
O7

Рисунок 3. Рентгенноргамма дифосфат никеля Ni2
P2
O7
Поглощение ИК-области обусловлено переходами между колебательными уровнями, отвечающими разной колебательной энергии функциональных групп. По ИК-спектрам поглощения конденсированных фосфатов может оказаться полезным не только для идентификации синтезированных соединений, но и для решения многих вопросов, связанных с их строением и химическими свойствами.
Образцы для записи спектров готовят, используя распространенную методику прессования вещества в бромистом калии (количество фосфата составляет 1-5 мг 700 мг KBr).
ИК спектры поглощения записывали в области 4000 – 400 см-1
с помощью спектрофотометра Sресоrd. Образцы готовили в виде таблеток, прессованных с КВr. Концентрация исследуемого вещества составляли 0,5 масс. %.
ИК-спектры поглощения конденсированных фосфатов расположены в порядке возрастания степени окисления металла, входящего в их состав, по аниону – в порядке возрастания степени конденсации аниона (Р2
О7
4-
– Р3
О10
5-
...[(3РО3
)n
]∞
→ Р3
О9
3-
– Р4
О12
4-
…), для кислых фосфатов – в порядке увеличения степени замещения атомов водорода металлом (например, Н3
Р2
О7
-
– Н2
Р2
О7
2-
– НР2
О7
3-
– Р2
О7
4-
) для ряда фосфатов, включающихкатионы металлов в одной и той же степени окисления – в порядке возрастания атомных масс металлов.
Конденсированные фосфаты, т.е. соли кислородных кислот фосфора (V), содержащих связи Р – О – Р, включают большую группу олигомерных и полимерных соединений с цепочными (ди-, три- и т.д.), циклическим (циклотри-, циклотетра- и т.д.) и разветвленным (ультрафосфаты) строением анионов.
Простейшим конденсированным фосфатным анионом является дифосфат – ион (пирофосфат - ион), образованный сочленением общими вершинами двух тетраэдров РО4
. При относительно простой структуре группы Р2
О7
ей присущи все основаны особенности химического строения конденсированных анионов, связанные с существованием и взаимным влиянием связей типа Р – О-
и Р – О(Р). Изменение характера этих связей находит свое отражение и в колебательных спектрах дифосфатов. Для интерпретации изменений валентных колебаний группы РО4
при переходе РО4
3-
– Р2
О7
4-
можно воспользоваться следующей схемой, которая с учетом вырождения устанавливает соответствие между 9 колебаниями группы Р2
О7
4-
:

Остальные три колебания группы Р2
О7
, отвечающие деформации угла РОР и внутренним вращениям вокруг связей Р – О(Р), лежат в низкочастотной области спектра.
Приведенная схема отражает экспериментально наблюдаемые изменения валентных колебаний Р – О при переходе РО4
→ Р2
О7
, которые прежде всего выражаются в том, что вместо одной частоты полносимметричного колебания появляются две. Эти две частоты описываются как vas
POP (более высокочастотная) и vs
POP (более низкочастотная), разрешены в ИК-спектрах для конфигурации Р2
О7
-группы C2
v
(угол РОР < 1800
), а для центосимметричной конфигурации D3
d
разрешена только vas
POP (угол РОР = 1800
). Расщепление частоты трижды вырожденного валентного колебания РО4
– v3 (F2) при переходе РО4→ Р2О7 мало по сравнению с расщеплением v1 (А1) на vs
и vas
POP, что объясняется значительно более высокой чувствительностью пульсационного колебания тетраэдра РО4
к взаимодействиям с колебаниями групп ппримыкающих к связи Р – О через атом кислорода.
Частотные интервалы проявляения валентных колебаний групп РО3
, РО2
, РОР и РО в спектрах дифосфатов и других конденсированных фосфатов по сравнению с vPO4
приведены на рисунке:

В спектре дифосфата полосы vs
POP может указывать на изогнутую или спрямленную конфигурацию мостика РОР. Поэтому представляет интерес возможность спектроскопической оценки величины угла РОР. Частоты vs
POP в спектрах большинства дифосфатов не выходят за пределы интервала 720 ± 20 см-4
. при увеличении угла РОР частота vs
POP должна понижаться, но повышение динамического коэффициента связи Р – О(Р), обычно сопровождающее увеличение угла РОР, может компенсировать изменение данной частоты. При этом оба фактора, и кинетический и динамический, действуют в одном направлении на vas
POP, приводя к повышению частоты от 900 см-1
в дифосфатах щелочных металлов до ~ 980 см-1
в дифосфатах магния и циркония. Использование величины ∆ = [vas
– vs
POP] / [vas
+ vs
POP] для суждения о величине угла РОР позволяет предполагать его постепенное уменьшение в ряду соединений Li4
P2
O7
, Na4
P2
O7
, K4
P2
O7
с ∆ = 12,0; 11,85 и 10,55% соответсвенно. Аналогичные изменения наблюдаются и в ряду дифосфатов щелочноземельных металлов от Mg2
P2
O7
∆ = 14,05% до Ва2
P2
O7
с ∆ = 11,9%.
Спектроскопическое исследование α – β-перехода М2
II
P2
O7
(MII
= Mg, Zn) позволяет определить не только конфигурацию группы Р2
О7
в α – форме, но и высказать предположение о вероятном механизме данного твердофазового превращения. По мнению авторов, он заключается не в статистическом распрямлении групп Р2
О7
, а связан с термическим возбуждением переброса атома О в связи Р – О – Р (и одновременно атомов О концевых групп РО3
) в эквивалентное относительна оси Р . . . Р положение, и при достаточно высокой температуре это приводит к статистической центросимметричности ионов Р2
О7
4-
.
На рис.6, 7, 8, 9, 10 показаны ИК – спектры синтезированных фосфатов цинка, никеля, железа и кобальта.
Основные колебательные частоты для Zn2
Р2
О7
· 5Н2
О находятся в дипазоне 3500 – 500 см-1
. Анализ ИК спектров исследуемых кристаллогидратов Zn2
Р2
О7
· 5Н2
О показал, что при комнатной температуре в области валентных колебаний ОН-групп молекул воды в диапозоне колебательной частоты 3500-3100 см-1
, наблюдаются полосы деформационного колебаний Н2
О.
На ИК – спектре дифосфата цинка наблюдаются так же полосы поглощения в области 1637,6 – 487,0 см-1
, соответствующие деформационным колебаниям антисимметричных и симметричных Р2
О7
4-
- групп.
В области (рис.1) 3182 – 2900см-1
в составе синтезированного вещества (Со2
Р2
О7
) наблюдается деформационные колебательные частоты 1403,0 – 596,1 см-1
, соответствуют основным антисиммеричным и симметричным колебательным частотам групп Р2
О7
4-
.
Результаты ИК спектра экспериментальных данных совпадают с литературными данными.
Таблица 4
Волновые числа (см-1
) максимумов полос поглощения в ИК спектрах Со2
Р2
О7
| Частота колебания, ν,см-1
. |
Полосы поглощения
|
| литературные данные |
синтезированный |
| 1535 |
1403,0 |
- |
| 1100 |
1111,4 |
νаs
(РО3
) |
| 738 |
734,7 |
νs
(РОР) |
| 588 |
596,1 |
δаs
РО |
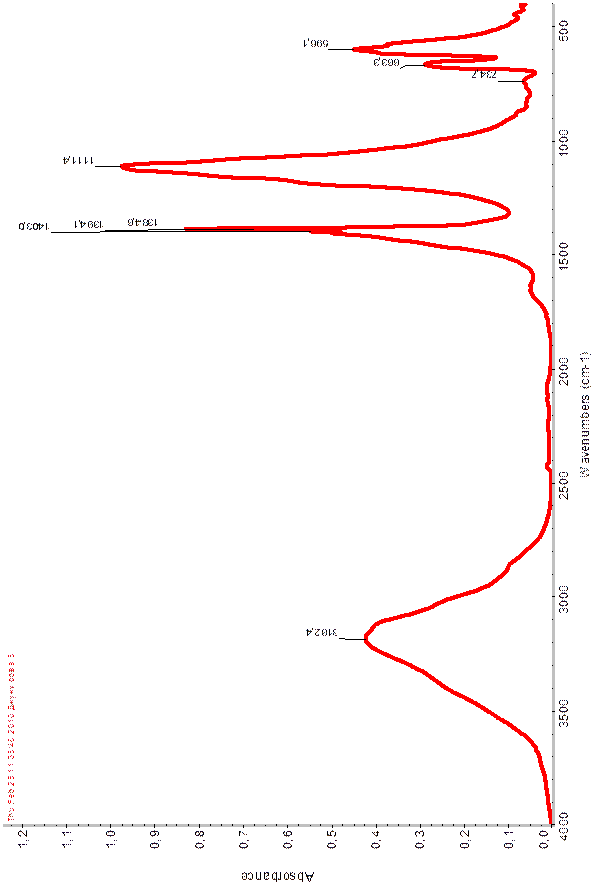
Рисунок 4. ИК – спектр Со2
Р2
О7
В области (рис.5, 6) 3127,8 – 1500см-1
в составе синтезированных веществ (Fe2
P2
O7
и Ni2
P2
O7
) наблюдаются широкие интенсивные полосы. Деформационные колебательные частоты соответствуют основным антисиммеричным и симметричным колебательным частотам групп Р2
О7
4-
.
Результаты ИК спектра экспериментальных данных совпадают с литературными данными.
Таблица 5
Волновые числа (см-1
) максимумов полос поглощения в ИК спектрах
Fe
2
P
2
O
7
и
Ni
2
P
2
O
7
| Частота колебания |
Частота поглощения,
ν,см-1
.
|
| литературные данные |
синтезированный |
| Fe2
P2
O7
|
| 1395 |
1384,6 |
| 1102 |
1095,0 |
νs
(РО3
) |
| 950 |
928,9 |
νаs
(РОР) |
| 601 |
595,5 |
νs
(РОР) |
| 535 |
520,7 |
δаs
(РО) |
| Ni2
P2
O7
|
| 1650 |
1614,6 |
δs
(Р2
О7
) |
| 1150 |
1161,0 |
νаs
(РО3
) |
| 1096 |
1080,2 |
νs
(РО3
) |
| 900 |
909,7 |
νаs
(РОР) |
| 746 |
750,0 |
νs
(РОР) |
| 496 |
490,9 |
δаs
(РО) |
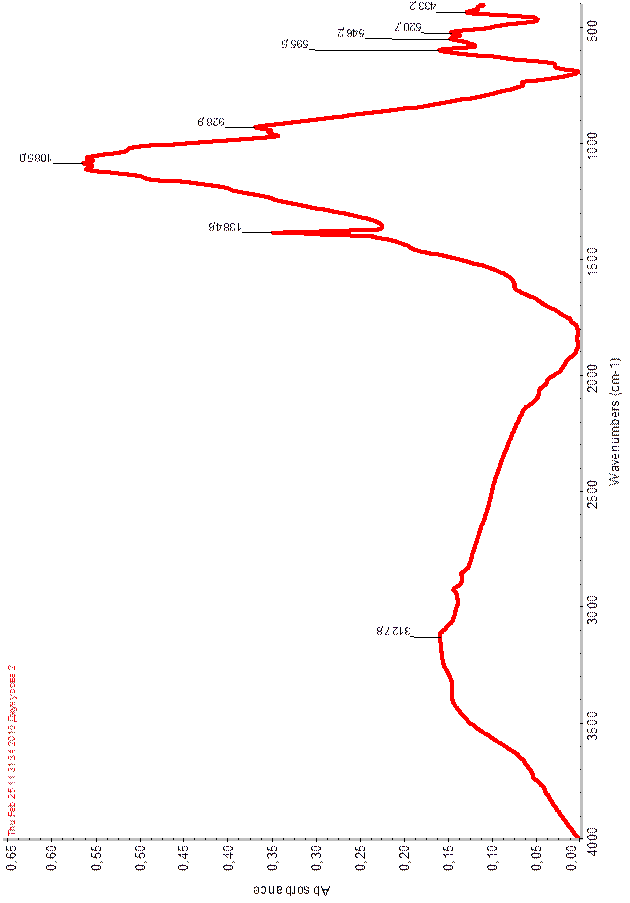
Рисунок 5. ИК-спектр Fe2
P2
O7

Рисунок 6. ИК – спектры Ni2
P2
O7
.
2.4.
Исследование сорбционных свойств
Статический метод
Этот метод, называемый многостадийным (каскадным) статическим процессом, является лабораторным, его применение ограничено вследствие большой затраты времени и возможности экспериментальных ошибок.
Как известно, вопросы сорбции тяжелых металоов неорганическими сорбентами представляют большой интерес. В работе [41] приводится результаты исследования кинетики сорбции марганца на Al(OH)3
, как одного из представителей тяжелых металлов с переменной степенью окисления. Методика, используемая в цитируемой работе, взята на основу.
Опыты по поглощению ионов Mn (II) проводились с растворами сульфата марганца, содержащих 60, 500, 1000 мкг/л ионов марганца, в статистических условиях [41].
Результаты по изучению сорбции ионов марганца (
II) дифосфатоми кобальта, железа и никеля, представленные на рисунках 11 – 15 показывают, что полное и практически полное поглощения сорбентом ионов марганца (II) из раствора солей марганца происходит при всех концентрациях раствора.
Наблюдающееся снижение содержания ионов Mn2+
в фильтрате обусловлено, на наш взгляд, следующим причинами. Ионы марганца (II) из раствора обмениваются с ионами железа (III) твердой фазы:
Fе2+
тв
+ Mn2+
р-р → Mn2+
тв
+ Fе2+
р-р
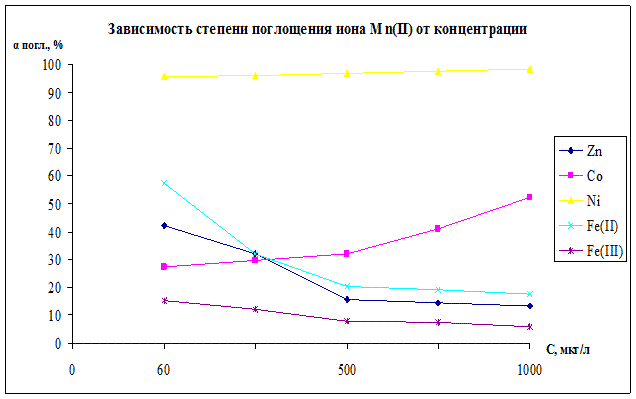
Рисунок 11.
На данном рисунке изображена зависимость степени поглощения иона марганца от концентрации раствора MnSO4
·5H2
O в статических условиях. В качестве сорбентов были использованы дифосфаты железа(II), кобальта, никеля. Кривые для Ni,Fe(II) показывают уменьшение значения степени поглощения иона марганца с увеличением концентрации раствора, при больших концентрациях степень поглощения приходит к стабильному значению и остается устойчивой. Но для кривой кобальта характерна другая зависимость, с увеличением концентрации наблюдается увеличение степени поглощения, что объясняется образованием сложных кобальто-марганцевых комплексов, наличие которых подтверждается результатами РФА, ИК-спектроскопии и химическим анализом. Ионы кобальта и марганца присутствуют и в твердой и в жидкой фазах, можно предположить ,что они образуют сложные комплексы, заметные на РФА в виде новой фазы. Максимальными значениями обладает кривая сорбции Ni, где αmax
= 98,5%.
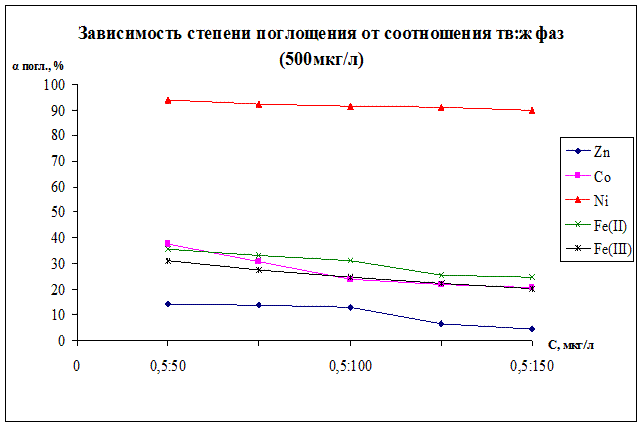
Рисунок 12.
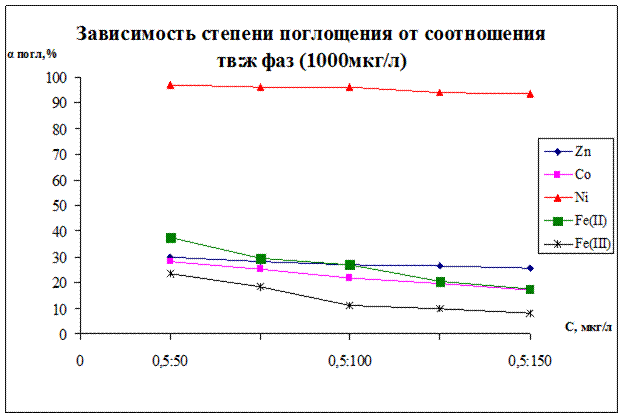
Рисунок 13.
На рисунках 12, 13 изображена зависимость степени поглощения иона марганца из раствора MnSO4
·5H2
O с различными концентрациями (500мкг/л,1000мкг/л) от соотношения твердой и жидкой фаз, в статических условиях. Данная зависимость является обратной для всех кривых, с увеличением объема жидкой фазы значения степени поглощения уменьшаются. Максимальными являются значения для кривой Ni,αmax
=97%. Большей концентрации сорбируемого раствора (1000мкг/л) соответствуют большие значения кривых поглощения.
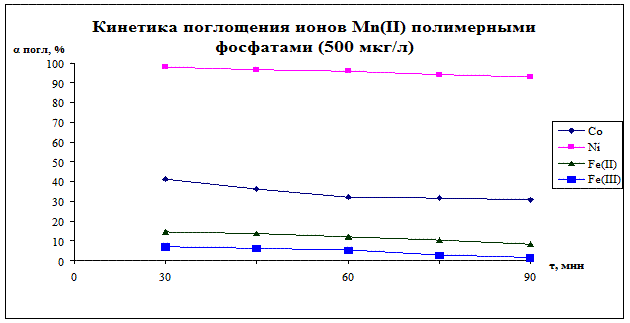
Рисунок 14.
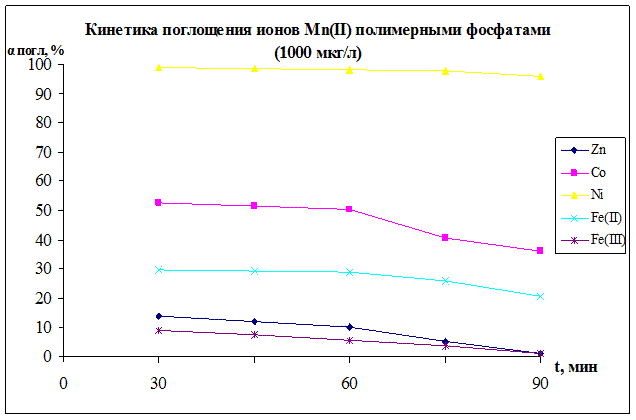
Рисунок 15.
На рисунках 14, 15 можно наблюдать зависимость степени поглощения иона марганца из раствора MnSO4
·5H2
O при различных концентрациях(500мкг/л,1000мкг/л) от времени. Кинетика процесса показывает, что процент поглощенного вещества убывает с увеличением времени, при τ =90мин значение начинает стремиться к нулю, что объясняется гидролизом ионита и изменением его структуры. Максимальные значения показывает кривая Ni, αmax
= 99%. Большей концентрации сорбируемого раствора (1000мкг/л) соответствуют большие значения αпогл
.
Таким образом, αпогл
исследованных выше конденсированных фосфатов наивысшей степенью поглощения обладает дифосфат никеля. Также определены факторы, влияющие сорбцию ионов Mn2+
: время τ, концентрация с и соотношение твердой и жидкой фазы сорбента и рабочего раствора т:ж. Кроме того заметные различия в значениях степени поглощения объясняются различной природой элементов и химизмом сорбционного процесса.
2.5. Химизм ионного процесса.
На основании проведенных исследований сделана попытка объяснения химизма происходящих процессов на дифосфатах Fe,Co,Ni(II).Процесс ионообмена в данном случае протекает по двум механизмам: по обменному и комплексообразованию. Обменный механизм характерен для ионов Fe(II), т.к. их ионные радиусы близки к сорбируемому иону Mn2+
[28].
Ионные радиусы
| Вид иона |
Радиус, нм |
| Mn2+
|
0,091 |
| Fe2+
|
0,080 |
| Co2+
|
0,078 |
| Ni2+
|
0,074 |
химически это можно выразить следующими уравнениями:
Fe2
P2
O7
+ MnSO4
· 5Н2
О = Mn2
P2
O7
+ FeSO4
+ 5Н2
О
Предположение об обменном механизме подтверждается экспериментальными данными атомно-абсорбционного анализа, содержание ионов Fe(II) в жидкой фазе после сорбции резко увеличивается, что говорит об их переходе в раствор. Чего нельзя сказать об ионах Ni и Co, содержание которых в жидкой фазе весьма невысокое (табл.6)
Таблица 6
Содержание иона марганца (
II
) после сорбции, %
| вещество
|
в твердой фазе
|
в растворе
|
| Ni2
P2
O7
|
77,2% |
22,5 % |
| Co2
P2
O7
|
43,4% |
56,5 % |
| Fe2
P2
O7
|
26,6% |
73,3 % |
Атом Ni имеет следующую конфигурацию внешних электронных уровней: d8
s0
, и способен к dsp2
гибридизации, за счет спаривания электронов на предвнешнем уровне может образовывать стабильные комплексы, с координационным числом равным 4, вероятно по следующей схеме:
Ni2
P2
O7
+ MnSO4
· 5Н2
О = Mn[NiP2
O7
] + NiSO4
+ 5Н2
О
Предположительно образуется два вида комплексов: а) Mn
[
NiP
2
O
7
]
 Mn Mn
OO
|| ||
О – Р – О – Р – O
 | | | |
OO

Ni
б) Ni
2
P
2
O
7
·
Mn
(
OH
)2
O
||
 HOO – P – OOH HOO – P – OOH
\ | /
MnOMn
/ | \
 OO – P – OO OO – P – OO
 || ||
O
Ni
Вторая структура имеет в своем составе (ОН) группы, по данным ИКС. При этом по данным РФА твердая фаза после сорбции перешла в аморфное фазу, что видно на рисунке 16.

Рисунок 16. Рентгенограмма Ni2
P2
O7
после сорбции
На ИК-спектре (Рис.17) наряду с полосами поглощения (РОР), (РО3
), (РО) группами обнаружена полоса поглощения (ОН) группы, в области диапазона 3300 см-1
.

Рисунок 17. ИК-спектр Ni2
P2
O7
после сорбции
Строение внешних уровней иона кобальта
отличается, он имеет нечетное количество d-электронов (d7
s0
), но в тоже время внешний уровень иона марганца также с нечетным числом электронов (d5
s0
), предположительно, взаимодействуя при комплексообразовании эти два иона образуют металл – металл связь Сo-Mn. Однако, по данным РФА, после сорбции основные соли (дифосфаты) являются самостоятельной фазой, т.к. процесс сорбции проходит не полностью их линии присутствуют в спектре, они находятся в одной области с образовавшейся новой фазой, подавляя друг друга, что затрудняет их идентификацию. Процесс ионообмена с кобальтом проходит частично по ионному механизму и частично с комплексообразованием, ионный радиус никеля меньше и тем самым его способность к компелксообразованию.
Выводы
1. Синтезированы и индетифицированы методами ИК-спектроскопии, РФА, химическими методами исследования конденсированные фосфаты цинка, железа, кобальта, никеля (Zn2
P2
O7
, Fe2
P2
O7
, Ni2
P2
O7
, Co2
P2
O7
).
2. Установлены зависимости сорбции ионов Mn(II) на полимерных фосфатах. Циклотетрафосфат железа не сорбирует ионы, из изученных дифосфатов Ni2
P2
O7
проявляет максимальную степень поглощения равную 98,5%.
3. Предложен химизм процесса сорбции ионов марганца дифосфатами железа, кобальта, никеля (Zn2
P2
O7
, Fe2
P2
O7
, Ni2
P2
O7
, Co2
P2
O7
), который происходит для Zn2+
, Fe2+
по ионным механизмам, а Co2+
, Ni2+
по комплексообразованию.
Использованная литература:
1. Куанышева Г.С., Макашева Г.Р., Джамансариева К.У. «Химия и основы модифицирования конденсированных фосфатов». Метод.пособие. Алматы, 2002г., 33 с.
2. Ван Везер «Фосфор и его соединения» М.: Изд.Инлит. 1962г. – 245 с.
3. Физико – химические исследования мнонмерных и полимерных фосфатов. Издательство Наука» Казахской СССР, А., 1987г.
4. Жданов Ю.В. «Химия и технология полифосфатов» - М.: Химия, 1979г., - 185 с.
5. Виноградова Н.В., Бондарь И.А., Демьянец Л.Н. и др. «Соединения редкоземельных элементов. Силикаты, германаты, фосфаты, арсенаты, ванадаты» М.: Наука, 1983г., - 122 с.
6. Корбридж Д. «Фосфор. Основы химии, биохимии, технологии» М.: Мир, 1982г. -с.119-125
7. Е.Е.Ергожин, М.К.Курманалиев Методическая разработка к спецпрактикуму «Иониты и ионный обмен», А., изд. КазГУ, 1988г., 37с.
8. Ключников Н.Г. «Руководство по неорганическому синтезу» - М.: Издательство «Химия»1965г.-311 с.
9. Ю.А Кокотов «Иониты и ионный обмен», Ленинград Химия, 1980. с. 100-106.
10. Ю.А. Кокотов, В.А.Пасечкин «Равновесие и кинетика ионного обмена» М.: Химия, 1970г., с.35-45
11. Куанышева Г.С., Макашева Г.Р. Методические указания к спецкурсу «Химия фосфорных удобрений». Алматы, КазГУ, 1989г. – с.6-20
12. Куанышева Г.С., Джамансариева К.У., Сдикова Г.Ж. «Термодинамические характеристики дифосфатов 3d-элементов» Вестник КазНУ, Сер. хим., 2005г, №2 (38), с.71-74
13. Е.Е.Ергожин «Высокопроницаемые иониты» А.: Наука, 1979г.
14. Констант З.А., Диндуне О.В. «Фосфаты двухвалентных металлов» Рига, Зинатне 1987г.- с.103-108
15. К.М.Солдадзе, В.Д.Копылова «Комплексообразующие иониты», М.: «Химия», 1980г. 220с.
16. М.Мархол «Ионообменники в аналитической химии», М.: Мир, 1985г., часть 1,2 с.25-190
17. Ч. Амфлетт «Неорганические иониты», М., Химия, 1966г. с. 85-95
18. Н.Г.Полянский, Г.В.Горбунов «Методы исследования ионитов», М.: Наука, 1976г., с.35-41
19. Е.Е.Ергожин, Е.Ж.Менлигазиев «Полифункциональные ионообменники» А.: Наука, 1986г.
20. Л.В.Зубанова, А.С.Тевлина, А.В.Даванков «Синтетические ионообменные материалы» - М.: Химия, 1973г.
21. Н.М.Селиванова, Н.Ю. Морозова, Л.Х.Кравченко, Т.И.Хожаинова «Термическая стойчивость пирофосфата цинка Zn2
Р2
О7
· 5Н2
О» Журнал неорган. Химии, 1975г., том 20, вып.3.
22. Krishnamachari N., Calvo C. The crystal structure of cobalt diphospate. – Acta Crystallogr. Sect.B, 1972, vol. 28B, N 9, p. 2883-2885.
23.Пахомов В.И. Некоторые вопросы кристаллохимии кислородных неорганических соединений фосфора (V). – Изв. АН СССР. Неорганич. материалы, 1977г., т. 13, № 8, с.1341-1352.
24. В.А.Урих, Третье всесоюзное совещание по фосфатам (тезисы), 3, Зинатне, Рига, 1971г., стр. 488-489.
25. А.Б.Бектуров, В.А.Урих, В.В.Тихонов, Д.З.Сератдезинов, В.А.Синяев, Известия АН СССР, Неорганическая материалы, 8, №2,
303, 1972г.
26. Lukaszewicz K. Crystal structure of α - Ni2
P2
O7
. Bull.Acad.Pol.Sci., 1967, Vol.15, №2, р.47-51
27. Тетеревков А.И., Печковский В.В. «Особенности термической диссоциации FePO4
» - ДАН БССР, 1974, т.18, № 5, с. 442-444.
28. «Краткий справочник физико-химических величин» Изд.8-е, перераб. Под ред. А.А.Равделя и А.М.Пономаревой. Л.: Химия, 1983г.: - 232 с.
29. Недома А. «Расшифровка рентгенограмм порошков».-Металлургия, 1975г., - 230 с.
30. Спицын В.И., Мартыненко Л.И. «Неорганическая химия», часть І, 1991г., - 245 с.
31. Щегров Л.Н. «Фосфаты двухвалентных металлов». Киев – Наукова думка. 1987г. –112 с.
32. Виноградов А.П. «Геохимия редких и рассеянных химических элементов в почвах» М.: Изд-во АН СССР, 1957г., - 218 с.
33. Бабенко Г.А. «Микроэлементы в сельском хозяйстве и медицине» К., 1963; - 125с.
34. Куанышева Г.С., Джамансариева К.У., Сдикова Г.Ж. «Изучение кинетики гидролитической деструкции дифосфатов 3d-элементов» Вестник КазНУ, Сер. хим., 2005г, №3 (39), с.156-159
35. Куанышева Г.С., Джамансариева К.У., Балгышева Б.Д., Макашева Г.Р. «Закономерности гидролитического расщепления линейных фосфатов s-элементов», Хабаршы вестник, 2002г. №3 (27), с.133-137
36. Тезисы и докладов 60-ой Республиканской научно-практической конференции молодых ученых и студентов по прикладным вопросам химии «Гидролитическая устойчивость циклотетрафосфатов 3d-элементов» Алматы, 2006г., 8с.
37. Ковальский В.В. «Геохимическая экология». М.: Наука, 1974., -195с.
38. ШкольникЯ., «Значение микроэлементов в жизни растений и в земледелии» Л., 1950г., - 251с.
39. Щегров Л.Н., Антрапцева Н.М. «Изучение взаимодействия смеси растворов сульфата цинка и кобальта с двухзамещенным фосфатом аммония», Журн.неорг.химии,1983г., 28 №10. с.2523-2528
40. Щегров Л.Н.«Образование двойных однозамещенных ортофосфатов Zn - Со», Укр.хим.журнал,1984, 50 №8. с.807-812.
41. С.М.Романова, З.С.Мусарипова, С.С.Крученко «Сорбция ионов марганца (II) гидроксидами железа и алюминия», Вестник КазГУ сер. Химическая, №3(15), А.: «Қазақ университеті», 1999г.
42. Бруцкус Е.Б., / тр. НИУИФ., т.137. с.101
43. Технология фосфорных и комплексных удобрений. М.: Химия, 1987г.
44. Маркина Т.Д. «Водные фосфаты алюминия, железа и хрома» Диссертация – Симферополь.
45. Сидоренко И., Журавлева Е.Д., Фордиенко В.И., Гринева Л.П., «Исследование комплексообразования ионов Fe3+
с фосфорной кислотой» ЖНХ., 1973. Т.18 В.5. с.:1270-1273
46. Филлатова Л.Н. «Комплексообразования железа (ІІІ) в концентрированных ортофасфатных растворах» ЖНХ., 1974. Т.19. В19. с.3064-3066
47. Rossee T., Kiesslich H. Chamatagraphisce analyse von phosphaten. /Anal. Chem.., 1976., p. 391-402.
48. «Атлас ИК-спектров фосфатов Моно-, дифосфаты, метафосфаты» под ред. Печковского В.В., М., Химия, 1971. с.: 248-256
49. Накамато К., «Инфракрасные спектры неорганических и координационных соединений», М.: Мир, 1966г.
50. Цымбал Е.П. «Физико-химическое исследование аморфной гидроокиси железа, осажденной водных и неводных растворов» Автореферат. Саратов, 1972г.
|