| ГОУ ВПО УГМА Росздрава
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2007г
ЛЕКЦИЯ № 7
Тема: Переваривание и всасывание углеводов. Обмен гликогена
Факультеты: лечебно-профилактический, медико-профилактический, педиатрический.
2 курс.
Углеводы
– это многоатомные спирты содержащие оксогруппу.
По количеству мономеров все углеводы делят на: моно-, ди-, олиго- и полисахариды.
Моносахариды по положению оксогруппы делятся альдозы и кетозы.
По количеству атомов углерода моносахариды делятся на триозы, тетрозы, пентозы, гексозы и т.д.
Функции углеводов
Моносахариды
– углеводы, которые не гидролизуются до более простых углеводов.
Моносахариды:
· выполняют энергетическую функцию (образование АТФ).
· выполняют пластическую функцию (участвуют в образовании ди-, олиго-, полисахаридов, аминокислот, липидов, нуклеотидов).
· выполняют детоксикационную функцию (производные глюкозы, глюкурониды, участвуют в обезвреживании токсичных метаболитов и ксенобиотиков).
· являются фрагментами гликолипидов (цереброзиды).
Дисахариды
– углеводы, которые гидролизуются на 2 моносахарида. У человека образуется только 1 дисахарид - лактоза. Лактоза синтезируется при лактации в молочных железах и содержится в молоке. Она:
· является источником глюкозы и галактозы для новорожденных;
· участвует в формировании нормальной микрофлоры у новорожденных.
Олигосахариды
– углеводы, которые гидролизуются на 3 - 10 моносахаридов.
Олигосахариды являются фрагментами гликопротеинов (ферменты, белки-транспортёры, белки-рецепторы, гормоны), гликолипидов (глобозиды, ганглиозиды). Они образуют на поверхности клетки гликокаликс.
Полисахариды
– углеводы, которые гидролизуются на 10 и более моносахаридов. Гомополисахариды выполняют запасающую функцию (гликоген – форма хранения глюкозы). Гетерополисахариды (ГАГ) являются структурным компонентом межклеточного вещества (хондроитинсульфаты, гиалуроновая кислота), участвуют в пролиферации и дифференцировке клеток, препятствуют свертыванию крови (гепарин).
Углеводы пищи, нормы и принципы нормирования их суточной пищевой потребности. Биологическая роль.
В пище человека в основном содержатся полисахариды — крахмал, целлюлоза растений, в меньшем количестве - гликоген животных. Источником сахарозы служат растения, особенно сахарная свёкла, сахарный тростник. Лактоза поступает с молоком млекопитающих (в коровьем молоке до 5% лактозы, в женском молоке — до 8%). Фрукты, мёд, соки содержат небольшое количество глюкозы и фруктозы. Мальтоза есть в солоде, пиве. Углеводы пищи являются для организма человека в основном источником моносахаридов, преимущественно глюкозы. Некоторые полисахариды: целлюлоза, пектиновые вещества, декстраны, у человека практически не перевариваются, в ЖКТ они выполняют функцию сорбента (выводят холестерин, желчные кислоты, токсины и д.р.), необходимы для стимуляции перистальтики кишечника и формирования нормальной микрофлоры.
Углеводы — обязательный компонент пищи, они составляют 75% массы пищевого рациона и дают более 50% необходимых калорий. У взрослого человека суточная потребность в углеводах 400г/сут, в целлюлозе и пектине до 10-15 г/сут. Рекомендуется употреблять в пищу больше сложных полисахаридов и меньше моносахаров.
Переваривание углеводов
Переваривание
это процесс гидролиза веществ до их ассимилируемых форм. Переваривание бывает: 1). Внутриклеточное (в лизосомах); 2). Внеклеточное (в ЖКТ): а). полостное (дистантное); б). пристеночное (контактное).
Переваривание углеводов в ротовой полости
(полостное)
В ротовой полости пища измельчается при пережёвывании и смачивается слюной. Слюна состоит на 99% из воды и обычно имеет рН 6,8. В слюне присутствует эндогликозидаза α-амилаза (α-1,4-гликозидаза),
расщепляющая в крахмале внутренние α-1,4-гликозидные связи с образованием крупных фрагментов — декстринов и небольшого количества мальтозы и изомальтозы. Необходим ион Cl-
.
Переваривание углеводов в желудке
(полостное)
Действие амилазы слюны прекращается в кислой среде (рН <4) содержимого желудка, однако, внутри пищевого комка активность амилазы может некоторое время сохраняться. Желудочный сок не содержит ферментов, расщепляющих углеводы, в нем возможен лишь незначительный кислотный гидролиз гликозидных связей.
Переваривание углеводов в тонком кишечнике
(полостное и пристеночное)
В двенадцатиперстной кишке кислое содержимое желудка нейтрализуется соком поджелудочной железы (рН 7,5—8,0 за счет бикарбонатов). С соком поджелудочной железы в кишечник поступает панкреатическая α-амилаза
. Эта эндогликозидаза гидролизует внутренние α-1,4-гликозидные связи в крахмале и декстринах с образованием мальтозы (2 остатка глюкозы, связанные α-1,4-гликозидной связью), изомальтозы (2 остатка глюкозы, связанные α-1,6-гликозидной связью) и олигосахаридов, содержащих 3—8 остатков глюкозы, связанных α-1,4- и α-1,6-гликозидными связями.
Переваривание мальтозы, изомальтозы и олигосахаридов происходит под действием специфических ферментов - экзогликозидаз, образующих ферментативные комплексы. Эти комплексы находятся на поверхности эпителиальных клеток тонкого кишечника и осуществляют пристеночное пищеварение.
Сахаразо-изомальтазный комплекс
состоит из 2 пептидов, имеет доменное строение. Из первого пептида образован цитоплазматический, трансмембранный (фиксирует комплекс на мембране энтероцитов) и связывающий домены и изомальтазная субъединица. Из второго - сахаразная субъединица. Сахаразная субъединица
гидролизует α-1,2-гликозидные связи в сахарозе, изомальтазная субъединица
- α-1,6-гликозидные связи в изомальтозе, α-1,4-гликозидные связи в мальтозе и мальтотриозе. Комплекса много в тощей кишке, меньше в проксимальной и дистальной частях кишечника.
Гликоамилазный комплекс
, содержит две каталитические субъединицы, имеющие небольшие различия в субстратной специфичности. Гидролизует α-1,4-гликозидные связи в олигосахаридах (с восстанавливающего конца) и в мальтозе. Наибольшая активность в нижних отделах тонкого кишечника.
β-Гликозидазный комплекс (лактаза)
гликопротеин, гидролизует β-1,4-гликозидные связи в лактозе. Активность лактазы зависит от возраста. У плода она особенно повышена в поздние сроки беременности и сохраняется на высоком уровне до 5-7-летнего возраста. Затем активность лактазы снижается, составляя у взрослых 10% от уровня активности, характерного для детей.Трегалаза
гликозидазный комплекс, гидролизует α-1,1-гликозидные связи между глюкозами в трегалозе — дисахариде грибов.Переваривание углеводов заканчивается образованием моносахаридов – в основном глюкозы, меньше образуется фруктозы и галактозы, еще меньше – маннозы, ксилозы и арабинозы.Всасывание углеводов
Моносахариды всасываются эпителиальными клетками тощей и подвздошной кишок. Транспорт моносахаридов в клетки слизистой оболочки кишечника может осуществляться путём диффузии (рибоза, ксилоза, арабиноза), облегчённой диффузии с помощью белков переносчиков (фруктоза, галактоза, глюкоза), и путем вторично-активного транспорта (галактоза, глюкоза). Вторично-активный транспорт галактозы и глюкозы из просвета кишечника в энтероцит осуществляется симпортом с Na+
. Через белок-переносчик Na+
двигается по градиенту своей концентрации и переносит с собой углеводы против их градиента концентраций. Градиент концентрации Na+
создаётся Nа+
/К+
-АТФ-азой. 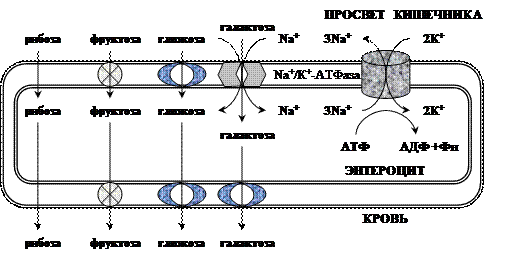  При низкой концентрации глюкозы в просвете кишечника она транспортируется в энтероцит только активным транспортом, при высокой концентрации - активным транспортом и облегчённой диффузией. Скорость всасывания: галактоза > глюкоза > фруктоза > другие моносахариды. Моносахариды выходят из энтероцитов в направлении кровеносного капилляра с помощью облегченной диффузии через белки-переносчики. При низкой концентрации глюкозы в просвете кишечника она транспортируется в энтероцит только активным транспортом, при высокой концентрации - активным транспортом и облегчённой диффузией. Скорость всасывания: галактоза > глюкоза > фруктоза > другие моносахариды. Моносахариды выходят из энтероцитов в направлении кровеносного капилляра с помощью облегченной диффузии через белки-переносчики. Нарушение переваривания и всасывания углеводов
Недостаточное переваривание и всасывание переваренных продуктов называют мальабсорбцией
. В основе мальабсорбции углеводов могут быть причины двух типов:
1). Наследственные и приобретенные дефекты ферментов, участвующих в переваривании
. Известны наследственные дефекты лактазы, α-амилазы, сахаразно-изомальтазного комплекса. Без лечения эти патологии сопровождаются хроническим дисбактериозом и нарушениями физического развития ребёнка.
Приобретённые нарушения переваривания могут наблюдаться при кишечных заболеваниях, например гастритах, колитах, энтеритах, после операций на ЖКТ.
Дефицит лактазы у взрослых людей может быть связан со снижением экспрессии гена лактазы, что проявляться непереносимостью молока - наблюдается рвота, диарея, спазмы и боли в животе, метеоризм. Частота этой патологии составляет в Европе 7—12%, в Китае — 80%, в Африке — до 97%.
2). Нарушение всасывания моносахаридов в кишечнике.
Нарушения всасывания могут быть следствием дефекта какого-либо компонента, участвующего в системе транспорта моносахаридов через мембрану. Описаны патологии, связанные с дефектом натрийзависимого белка переносчика глюкозы.
Синдром мальабсорбции сопровождается осмотической диареей, усилением перистальтики, спазмами, болями, а также метеоризмом. Диарею вызывают нерасщеплённые дисахариды или невсосавшиеся моносахариды в дистальных отделах кишечника, а также органические кислоты, образованные микроорганизмами при неполном расщеплении углеводов.
Транспорт глюкозы из крови в клетки
Глюкоза поступает из кровотока в клетки путём облегчённой диффузии с помощью белков-переносчиков - ГЛЮТов. Глюкозные транспортёры ГЛЮТы имеют доменную организацию и обнаружены во всех тканях. Выделяют 5 типов ГЛЮТов:• ГЛЮТ-1 - преимущественно в мозге, плаценте, почках, толстом кишечнике;• ГЛЮТ-2 - преимущественно в печени, почках, β-клетках поджелудочной железы, энтероцитах, есть в эритроцитах. Имеет высокую Км; • ГЛЮТ-3 - во многих тканях, включая мозг, плаценту, почки. Обладает большим, чем ГЛЮТ-1, сродством к глюкозе;
• ГЛЮТ-4 - инсулинзависимый, в мышцах (скелетной, сердечной), жировой ткани;• ГЛЮТ-5 - много в клетках тонкого кишечника, является переносчиком фруктозы. ГЛЮТы, в зависимости от типа, могут находиться преимущественно как в плазматической мембране, так и в цитозольных везикулах. Трансмембранный перенос глюкозы происходит только тогда, когда ГЛЮТы находятся в плазматической мембране. Встраивание ГЛЮТов в мембрану из цитозольных везикул происходит под действием инсулина. При снижении концентрации инсулина в крови эти ГЛЮТы снова перемещаются в цитоплазму. Ткани, в которых ГЛЮТы без инсулина почти полностью находятся в цитоплазме клеток (ГЛЮТ-4, и в меньшей мере ГЛЮТ-1), оказываются инсулинзависимыми (мышцы, жировая ткань), а ткани, в которых ГЛЮТы преимущественно находятся в плазматической мембране (ГЛЮТ-3) - инсулиннезависимыми.
Известны различные нарушения в работе ГЛЮТов. Наследственный дефект этих белков может лежать в основе инсулинонезависимого сахарного диабета.
Метаболизм моносахаридов в клетке
После всасывания в кишечнике глюкоза и другие моносахариды поступают в воротную вену и далее в печень. Моносахариды в печени превращаются в глюкозу или продукты её метаболизма. Часть глюкозы в печени депонируется в виде гликогена, часть идет на синтез новых веществ, а часть через кровоток, направляется в другие органы и ткани. При этом печень поддерживает концентрацию глюкозы в крови на уровне 3,3-5,5 ммоль/л.
Фосфорилирование и дефосфорилирование моносахаридов
В клетках глюкоза и другие моносахариды с использованием АТФ фосфорилируются до фосфорных эфиров: глюкоза + АТФ → глюкоза-6ф + АДФ. Для гексоз эту необратимую реакцию катализирует фермент гексокиназа
, которая имеет изоформы: в мышцах - гексокиназа II, в печени, почках и β-клетках поджелудочной железы - гексокиназа IV (глюкокиназа), в клетках опухолевых тканей - гексокиназа III. Фосфорилирование моносахаридов приводит к образованию реакционно-способных соединений (реакция активации), которые не способны покинуть клетку т.к. нет соответствующих белков-переносчиков. Фосфорилирование уменьшает количество свободной глюкозы в цитоплазме, что облегчает ее диффузию из крови в клетки.
Гексокиназа
II
фосфорилирует D-глюкозу, и с меньшей скоростью, другие гексозы. Обладая высоким сродством к глюкозе (Кm <0,1 ммоль/л), гексокиназа II обеспечивает поступление глюкозы в ткани даже при низкой концентрации глюкозы в крови. Так как гексокиназа II ингибируется глюкозо-6-ф (и АТФ/АДФ), глюкоза поступает в клетку только по мере необходимости.
Глюкокиназа
(гексокиназа IV) имеет низкое сродство к глюкозе (Кm - 10 ммоль/л), активна в печени (и почках) при повышении концентрации глюкозы (в период пищеварения). Глюкокиназа не ингибируется глюкозо-6-фосфатом, что дает возможность печени без ограничений удалять излишки глюкозы из крови.
Глюкозо-6-фосфатаза
катализирует необратимое отщепление фосфатной группы гидролитическим путём в ЭПР: Глюкозо-6-ф + Н2
О → Глюкоза + Н3
РО4
, есть только в печени, почках и клетках эпителия кишечника. Образовавшаяся глюкоза способна диффундировать из этих органов в кровь. Таким образом, глюкозо-6-фосфатаза печени и почек позволяет повышать низкий уровень глюкозы в крови.
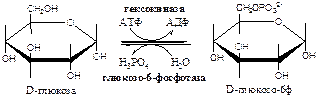
Метаболизм глюкозо-6-фосфата
Глюкозо-6-ф может использоваться клетке в различных превращениях, основными из которых являются: катаболизм с образованием АТФ, синтез гликогена, липидов, пентоз, полисахаридов и аминокислот.
МЕТАБОЛИЗМ ГЛИКОГЕНА
Многие ткани в качестве резервной формы глюкозы синтезируют гликоген. Синтез и распад гликогена в печени поддерживают гомеостаз глюкозы в крови.
Гликоген
— разветвлённый гомополисахарид глюкозы с массой >107
Да (50000 остатков глюкозы), в котором остатки глюкозы соединены в линейных участках α-1,4-гликозидной связью. В точках ветвления, примерно через каждые 10 остатков глюкозы, мономеры соединены α-1,6-гликозидными связями. Гликоген, водонерастворим, хранится в цитозоле клетки в форме гранул диаметром 10-40 нм. Гликоген депонируется главным образом в печени (до 5%) и скелетных мышцах (до 1%). В организме может содержаться от 0 до 450 г гликогена.
Разветвлённая структура гликогена способствует работе ферментов, отщепляющих или присоединяющих мономеры.
Синтез гликогена (гликогеногенез)
Гликоген синтезируется с затратой энергии в период пищеварения (через 1—2 ч после приёма углеводной пищи).
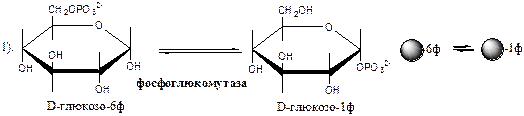
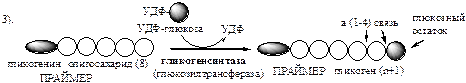
Синтез гликогена осуществляется путём удлинения уже имеющейся молекулы полисахарида, называемой «затравка
», или «праймер
». В состав праймера может входить белок гликогенин, в котором к Тир присоединен олигосахарид (примерно из 8 остатков глюкозы). Глюкозные остатки переносятся гликогенсинтазой на нередуцирующий конец олигосахарида и связываются α-1,4-гликозидными связями.
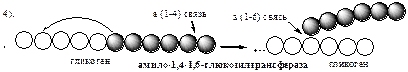
При удлинении линейного участка примерно до 11 глюкозных остатков, фермент ветвления переносит её концевой блок, содержащий 6—7 остатков, на внутренний остаток глюкозы этой или другой цепи с образованием α-1,6-гликозидной связи. Новая точка ветвления образуется на расстоянии не менее 4 остатков от любой уже существующей точки ветвления.
Распад гликогена (гликогенолиз)
Распад гликогена происходит путем последовательного отщепления глюкозо-1-ф в ответ на повышение потребности организма в глюкозе. Реакцию катализирует гликогенфосфорилаза:

Гликогенфосфорилаза
состоит из 2 идентичных субъединиц (94500 Да). Неактивная форма обозначается b, активная - a. Активируется киназой фосфорилазы
b
путем
фосфорилирования каждой субъединицы по серину в 14 положении.
Гликогенфосфорилаза расщепляет фосфоролизом α-1,4-гликозидные связи, до тех пор, пока до точки ветвления не остается 4 остатка глюкозы.
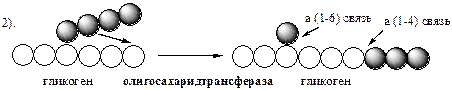
Инактивация гликогенфосфорилазы происходит при дефосфорилировании с участием специфической фосфатазы фосфорилазы (фосфопротеинфосфотазы ФПФ).
Удаление ветвления осуществляет деветвящий фермент
. Он обладает трансферазной и гликозидазной активностями. Трасферазная часть (олигосахаридтрансфераза
) переносит три оставшихся до точки ветвления глюкозных остатка на нередуцирующий конец соседней цепи, удлиняя её для фосфорилазы.
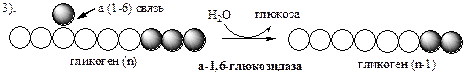
Гликозидазная часть (α-1,6-глюкозидаза
) гидролизует α-1,6-гликозидную связь, отщепляя глюкозу.
Глюкозо-1-ф изомеризуется в глюкозо-6-ф фосфоглюкомутазой.
Регуляция метаболизма гликогена в печени

     
 
    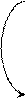 
Регуляция метаболизма гликогена в мышцах
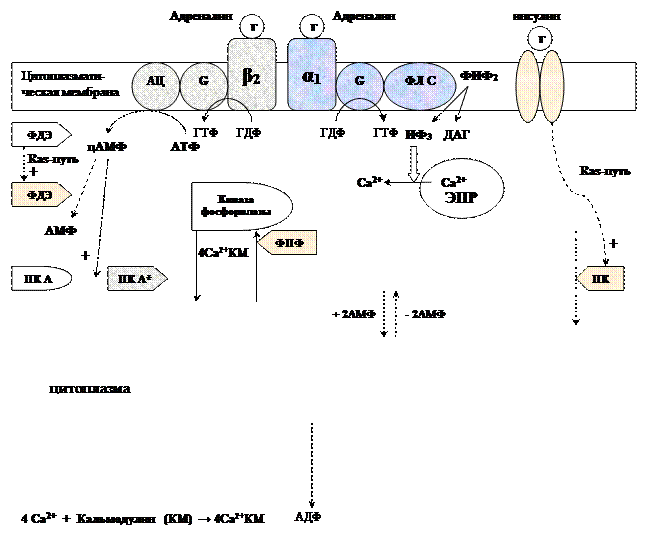 Метаболизм гликогена контролируется гормонами (в печени - инсулином, глюкагоном, адреналином; в мышцах - инсулином и адреналином), которые регулируют фосфорилирование /дефосфорилирование 2 ключевых ферментов гликогенсинтазы и гликогенфосфорилазы.
При недостаточном уровне глюкозы в крови выделяется гормон глюкагон, в крайних случаях – адреналин. Они стимулируют фосфорилирование гликогенсинтазы (она инактивируется) и гликогенфосфорилазы (она активируется). При повышении уровня глюкозы в крови выделяется инсулин, он стимулирует дефосфорилирование гликогенсинтазы (она активируется) и гликогенфосфорилазы (она инактивируется). Кроме того, инсулин индуцирует синтез глюкокиназы, тем самым, ускоряя фосфорилирование глюкозы в клетке. Всё это приводит к тому, что инсулин стимулирует синтез гликогена, а адреналин и глюкагон – его распад.
В печени существует и аллостерическая регуляция гликогенфосфорилазы: ее ингибирует АТФ и глюкозо-6ф, а активирует АМФ.
Нарушения обмена гликогена
Гликогеновые болезни
— группа наследственных нарушений, в основе которых лежит снижение или отсутствие активности ферментов, катализирующих реакции синтеза или распада гликогена, либо нарушение регуляции этих ферментов.
Гликогенозы
— заболевания, обусловленные дефектом ферментов, участвующих в распаде гликогена. Они проявляются или необычной структурой гликогена, или его избыточным накоплением в печени, сердечной или скелетных мышцах, почках, лёгких и других органах.
В настоящее время гликогенозы делят на 2 группы: печёночные и мышечные.
Печёночные формы гликогенозов
ведут к нарушению использования гликогена для поддержания уровня глюкозы в крови. Поэтому общий симптом для этих форм — гипогликемии в постабсорбтивный период.
Болезнь Гирке
(тип I) отмечают наиболее часто. Причина — наследственный дефект глюкозо-6-фосфатазы — фермента, обеспечивающего выход глюкозы в кровоток после её высвобождения из гликогена клеток печени и почек. Клетки печени и извитых канальцев почек заполнены гликогеном, печень и селезенка увеличены, у больных опухлое лицо - «лицо китайской куклы». Болезнь проявляется гипогликемией, гипертриацилглицеролемией, гиперурикемией, ацидоз.
1). В гепатоцитах: ↑глюкозо-6-ф → ↑ПВК, ↑лактат (ацидоз), ↑рибозо-5-ф. ↑рибозо-5-ф→ ↑пуринов→ ↑ мочевая кислота
2). В крови: ↓глюкоза →↓инсулин/глюкагон→: а) ↑липолиз жировой ткани → ↑ЖК в крови.
б). ↓ЛПЛ жировой ткани → ↑ТАГ в крови.
Лечение - диета по глюкозе, частое кормление.
Болезнь Кори
(тип III) распространена, 1/4 всех печёночных гликогенозов. Накапливается разветвленный гликоген, так как дефектен деветвящий фермент. Гликогенолиз возможен, но в незначительном объёме. Лактоацидоз и гиперурикемия не отмечаются. Болезнь отличается более лёгким течением чем болезнь Гирке.
Мышечные формы гликогенозов
характеризуются нарушением в энергоснабжении скелетных мышц. Эти болезни проявляются при физических нагрузках и сопровождаются болями и судорогами в мышцах, слабостью и быстрой утомляемостью.
Болезнь МакАрдла
(тип V) — аутосомно-рецессивная патология, отсутствует в скелетных мышцах активность гликогенфосфорилазы. Накопление в мышцах гликогена аномальной структуры.
Агликогенозы
Агликогеноз
(гликогеноз 0 по классификации) — заболевание, возникающее в результате дефекта гликогенсинтазы. В печени и других тканях больных наблюдают очень низкое содержание гликогена. Это проявляется резко выраженной гипогликемией в постабсорбтивном периоде. Характерный симптом — судороги, проявляющиеся особенно по утрам. Болезнь совместима с жизнью, но больные дети нуждаются в частом кормлении.
ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2007 г
ЛЕКЦИЯ № 8
Тема: Катаболизм глюкозы. Гликолиз
Основные пути катаболизма глюкозы
Катаболизм глюкозы в клетке может проходить как в аэробных, так и в анаэробных условиях, его основная функция - это синтез АТФ.
Аэробное окисление глюкозы
В аэробных условиях глюкоза окисляется до СО2
и Н2
О. Суммарное уравнение:
С6
Н12
О6
+ 6О2
→ 6СО2
+ 6Н2
О + 2880 кДж/моль.
Этот процесс включает несколько стадий:
1. Аэробный гликолиз
. В нем происходит окисления 1 глюкозы до 2 ПВК, с образованием 2 АТФ (сначала 2 АТФ затрачиваются, затем 4 образуются) и 2 НАДН2
;
2. Превращение 2 ПВК в 2 ацетил-КоА с выделением 2 СО2
и образованием 2 НАДН2
;
3. ЦТК.
В нем происходит окисление 2 ацетил-КоА с выделением 4 СО2
, образованием 2 ГТФ (дают 2 АТФ), 6 НАДН2
и 2 ФАДН2
;
4. Цепь окислительного фосфорилирования.
В ней происходит окисления 10 (8) НАДН2
, 2 (4) ФАДН2
с участием 6 О2
, при этом выделяется 6 Н2
О и синтезируется 34 (32) АТФ.
В результате аэробного окисления глюкозы образуется 38 (36) АТФ, из них: 4 АТФ в реакциях субстратного фосфорилирования, 34 (32) АТФ в реакциях окислительного фосфорилирования. КПД аэробного окисления составит 65%.
Анаэробное окисление глюкозы
Катаболизм глюкозы без О2
идет в анаэробном гликолизе и ПФШ (ПФП).
· В ходе анаэробного гликолиза
происходит окисления 1 глюкозы до 2 молекул молочной кислоты с образованием 2 АТФ (сначала 2 АТФ затрачиваются, затем 4 образуются). В анаэробных условиях гликолиз является единственным источником энергии. Суммарное уравнение: С6
Н12
О6
+ 2Н3
РО4
+ 2АДФ → 2С3
Н6
О3
+ 2АТФ + 2Н2
О.
· В ходе ПФП
из глюкозы образуются пентозы и НАДФН2
. В ходе ПФШ
из глюкозы образуются только НАДФН2
.
ГЛИКОЛИЗ
Гликолиз – главный путь катаболизма глюкозы (а также фруктозы и галактозы). Все его реакции протекают в цитозоле.
Аэробный гликолиз
- это процесс окисления глюкозы до ПВК, протекающий в присутствии О2
.
Анаэробный гликолиз
– это процесс окисления глюкозы до лактата, протекающий в отсутствии О2
.
Анаэробный гликолиз отличается от аэробного только наличием последней 11 реакции, первые 10 реакций у них общие.
Этапы гликолиза
В любом гликолизе можно выделить 2 этапа:
- 1 этап подготовительный, в нем затрачивается 2 АТФ. Глюкоза фосфорилируется и расщепляется на 2 фосфотриозы;
- 2 этап, сопряжён с синтезом АТФ. На этом этапе фосфотриозы превращаются в ПВК. Энергия этого этапа используется для синтеза 4 АТФ и восстановления 2НАДН2
, которые в аэробных условиях идут на синтез 6 АТФ, а в анаэробных условиях восстанавливают ПВК до лактата.
Энергетический баланс гликолиза
Таким образом, энергетический баланс аэробного гликолиза:
8АТФ = -2АТФ + 4АТФ + 6АТФ (из 2НАДН2
)
Энергетический баланс анаэробного гликолиза:
2АТФ = -2АТФ + 4АТФ
Общие реакции аэробного и анаэробного гликолиза
1. Гексокиназа
(гексокиназа II, АТФ: гексозо-6-фосфотрансфераза) в мышцах фосфорилирует в основном глюкозу, меньше – фруктозу и галактозу.
Кm<0,1 ммоль/л. Ингибитор глюкозо-6-ф, АТФ. Активатор адреналин. Индуктор инсулин.
Глюкокиназа
(гексокиназа IV, АТФ: глюкозо-6-фосфотрансфераза) фосфорилирует глюкозу. Кm - 10 ммоль/л, активна в печени, почках. Не ингибируется глюкозо-6-ф. Индуктор инсулин. Гексокиназы осуществляют фосфорилирование гексоз.
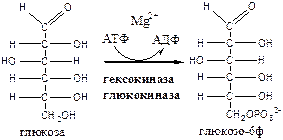
2.
Фосфогексозоизомераза
(глюкозо-6ф-фруктозо-6ф-изомераза) осуществляет альдо-кетоизомеризацию открытых форм гексоз.
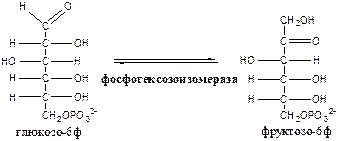
3.
Фосфофруктокиназа 1
(АТФ: фруктозо-6ф-1-фосфотрансфераза) осуществляет фосфорилирование фруктозы-6ф. Реакция необратима и самая медленная из всех реакций гликолиза, определяет скорость всего гликолиза. Активируется: АМФ, фруктозо-2,6-дф (мощный активатор, образуется с участием фосфофруктокиназы 2 из фруктозы-6ф), фруктозо-6-ф, Фн. Ингибируется: глюкагоном, АТФ, НАДН2
, цитратом, жирными кислотами, кетоновыми телами. Индуктор реакции инсулин.

4.
Альдолаза А
(фруктозо-1,6-ф: ДАФ-лиаза). Альдолазы действуют на открытые формы гексоз, имеют 4 субъединицы, образуют несколько изоформ. В большинстве тканей содержится Альдолаза А. В печени и почках – Альдолаза В.
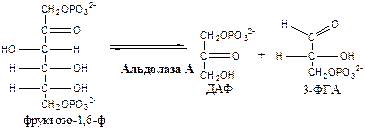
5. Фосфотриозоизомераза
(ДАФ-ФГА-изомераза).

6.
3-ФГА дегидрогеназа
(3-ФГА: НАД+
оксидоредуктаза (фосфорилирующая)) состоит из 4 субъединиц. Катализирует образование макроэргической связи в 1,3-ФГК и восстановление НАДН2
, которые используются в аэробных условиях для синтеза 8 (6) молекул АТФ.
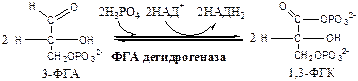
7.
Фосфоглицераткиназа
(АТФ: 3ФГК-1-фосфотрансфераза). Осуществляет субстратное фосфорилирование АДФ с образованием АТФ.
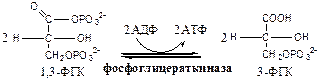
В следующих реакциях низкоэнергетический фосфоэфир переходит в высокоэнергетический фосфат.
8.
Фосфоглицератмутаза
(3-ФГК-2-ФГК-изомераза) осуществляет перенос фосфатного остатка в ФГК из положения 3 положение 2.
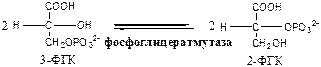
9.
Енолаза
(2-ФГК: гидро-лиаза) отщепляет от 2-ФГК молекулу воды и образует высокоэнергетическую связь у фосфора. Ингибируется ионами F-
.
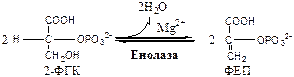
10.
Пируваткиназа
(АТФ: ПВК-2-фосфотрансфераза) осуществляет субстратное фосфорилирование АДФ с образованием АТФ. Активируется фруктозо-1,6-дф, глюкозой. Ингибируется АТФ, НАДН2
, глюкагоном, адреналином, аланином, жирными кислотами, Ацетил-КоА. Индуктор: инсулин, фруктоза.
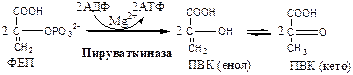
Образующаяся енольная форма ПВК затем неферментативно переходит в более термодинамически стабильную кетоформу. Данная реакция является последней для аэробного гликолиза.
Дальнейший катаболизм 2 ПВК и использование 2 НАДН2
зависит от наличия О2
.
Реакция анаэробного гликолиза
В анаэробных условиях ПВК, подобно О2
в дыхательной цепи, обеспечивает регенерацию НАД+
из НАДН2
, что необходимо для продолжения реакций гликолиза. ПВК при этом превращается в молочную кислоту. Реакция протекает в цитоплазме с участием лактатдегидрогеназы (ЛДГ).
11.
Лактатдегидрогеназа
(лактат: НАД+
оксидоредуктаза). Стоит из 4 субъединиц, имеет 5 изоформ.
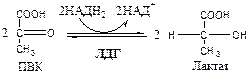
Лактат не является конечным продуктом метаболизма, удаляемым из организма. Из анаэробной ткани лактат переноситься кровью в печень, где превращаясь в глюкозу (Цикл Кори), или в аэробные ткани (миокард), где превращается в ПВК и окисляется до СО2
и Н2
О.
Катаболизм ПВК в митохондриях
В аэробных условиях ПВК и водороды с НАДН2
транспортируются в матрикс митохондрий. ПВК самостоятельно не проходит внутреннюю мембрану митохондрий, перенос ее через мембрану осуществляется вторично-активным транспортом симпортом с Н+
. ПВК в митохондриях используется в 2 реакциях:
1. Пируватдегидрогеназный комплекс
(ПВК: НАД+
оксидорудуктаза (декарбоксилирующая)) содержит 3 фермента и 5 коферментов: а) Пируватдекарбоксилаза содержит (Е1
) 120 мономеров и кофермент ТПФ; б) Дигидролипоилтрансацилаза (Е2
) содержит 180 мономеров и коферменты липоамид и HSКоА; в) Дигидролипоилдегидрогеназа (Е3
) содержит 12 мономеров и коферменты ФАД и НАД. Пируват ДГ комплекс
осуществляет окислительное декарбоксилирование ПВК с образованием Ацетил-КоА. Активатор: HSКоА, НАД+
, АДФ. Ингибитор: НАДН2
, АТФ, Ацетил-КоА, жирные кислоты, кетоновые тела. Индуктор инсулин.
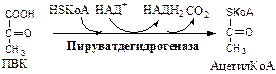
Механизм работы Пируват ДГ комплекса. Процесс проходит 5 стадий:
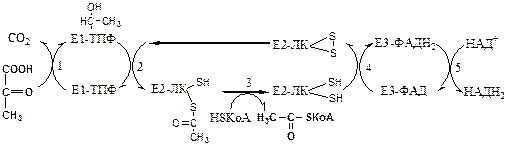
Далее Ацетил-КоА поступает в ЦТК, где он окисляется до 2 СО2
с образованием 1 ГТФ, восстановлением 3 НАДН2
и 1 ФАДН2
.
2. Пируваткарбоксилаза
(ПВК: СО2
-синтетаза (АТФ → АДФ + Фн)) сложный олигомерный фермент, содержит биотин. Карбоксилирует ПВК до ЩУК. Пополняющая реакция, по мере необходимости добавляет ЩУК в ЦТК. Активатор: Ацетил-КоА.
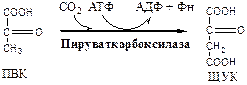
Челночные системы
В аэробных условиях О2
обеспечивает регенерацию НАД+
из НАДН2
, что необходимо для продолжения реакции гликолиза (НАД+
субстрат 3-ФГА ДГ).
Так как внутренняя мембрана митохондрий непроницаема для НАДН2
, восстановленный в гликолизе НАДН2
, передает свои водороды на дыхательную цепь митохондрий с помощью специальных систем, называемых «челночными». Известны 2 челночные системы: малат-аспартатная и глицерофосфатная.
1. Малат-аспартатный челнок
является универсальным, работает в печени, почках, сердце.
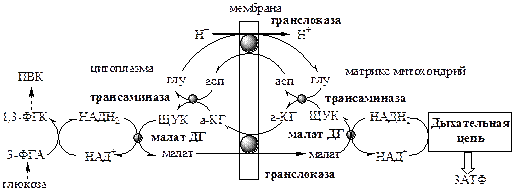 2. 2.
Глицерофосфатный челночный механизм.
Работает в белых скелетных мышцах
, мозге, в жировой ткани, гепатоцитах
.
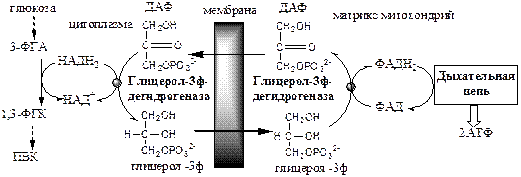
Малат-аспартатный челнок энергетически более эффективен, так как передаёт водород в дыхательную цепь через митохондриальный НАД, соотношение Р/О равно 3, синтезируется 3 АТФ.
В глицерофосфатный челнок передаёт водород в дыхательную цепь через ФАД на KoQ, соотношение Р/О равно 2, синтезируется 2 АТФ.
Пластическое значение катаболизма глюкозы
При катаболизме глюкоза может выполнять пластические функции. Метаболиты гликолиза используются для синтеза новых соединений. Так, фруктозо-6ф и 3-ФГА участвуют в образовании рибозо-5-ф (компонент нуклеотидов); 3-фосфоглицерат может включаться в синтез аминокислот, таких как серии, глицин, цистеин. В печени и жировой ткани Ацетил-КоА используется при биосинтезе жирных кислот, холестерина, а ДАФ для синтеза глицерол-3ф.
Регуляция гликолиза
Эффект Пастера
– снижение скорости потребления глюкозы и накопления лактата в присутствии кислорода.
Эффекта Пастера объясняется наличием конкуренции между ферментами аэробного (ПВК ДГ, ПВК карбоксилаза, ферменты цепи окислительного фосфорилирования) и анаэробного (ЛДГ) пути окисления за общий метаболит ПВК
и кофермент НАДН2
.
· Без О2
митохондрии не потребляют ПВК и НАДН2
, в результате их концентрация в цитоплазме повышается и они идут на образование лактата. Так как анаэробный гликолиз дает из 1 глюкозы только 2 АТФ, для образования достаточного количества АТФ необходимо много глюкозы (в 19 раз больше чем в аэробных условиях).
· В присутствии О2
, митохондрии выкачивают ПВК и НАДН2
из цитоплазмы, прерывая реакцию образования лактата. При аэробном окислении из 1 глюкозы образуется 38 АТФ, соответственно для образования достаточного количества АТФ необходимо мало глюкозы (в 19 раз меньше чем в анаэробных условиях).
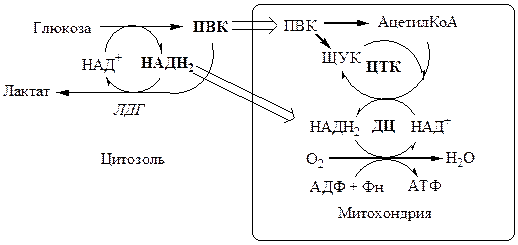
МЕТАБОЛИЗМ ФРУКТОЗЫ И ГАЛАКТОЗЫ
Фруктоза и галактоза наряду с глюкозой используются для получения энергии или синтеза веществ: гликогена, ТГ, ГАГ, лактозы и др.
Метаболизм фруктозы
Значительное количество фруктозы, образующееся при расщеплении сахарозы, превращается в глюкозу уже в клетках кишечника. Часть фруктозы поступает в печень.
Метаболизм фруктозы в клетке начинается с реакции фосфорилирования:
1.
Фруктокиназа
(АТФ: фруктоза-1-фосфотрансфераза) фосфорилирует только фруктозу, имеет к ней высокое сродство. Содержится в печени, почках, кишечнике. Инсулин не влияет на ее активность.
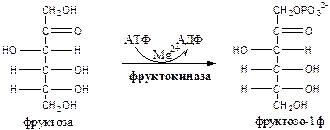
2. Альдолаза В
(фруктозо: ГА-лиаза) есть в печени, расщепляет фруктозо-1ф (фруктозо-1,6ф) до глицеринового альдегида (ГА) и диоксиацетонфосфата (ДАФ).
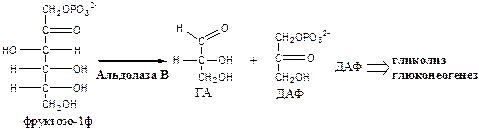
3.
Триозокиназа
(АТФ: ГА-3-фосфотрансфераза). Много в печени.
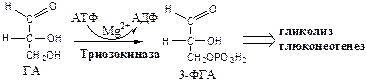
ДАФ и ГА, полученные из фруктозы, включаются в печени главным образом в глюконеогенез. Часть ДАФ может восстанавливаться до глицерол-3-ф и участвовать в синтезе ТГ.
Нарушения метаболизма фруктозы
Причиной нарушения метаболизма фруктозы является дефект 3 ферментов: фруктокиназы, альдолазы В, триозокиназы.
Доброкачественная эссенциальная фруктозурия
связана с недостаточностью
фруктокиназы
,
клинически не проявляется. Фруктоза накапливается в крови и выделяется с мочой, где её можно обнаружить лабораторными методами. Частота 1:130 000.
Наследственная непереносимость фруктозы
частая патология, возникает при генетически дефекте альдолазы В
(аутосомно-рецессивная форма). Она проявляется, когда в рацион добавляют фрукты, соки, сахарозу. После приёма пищи, содержащей фруктозу возникает рвота, боли в животе, диарея, гипогликемия и даже кома и судороги
. У маленьких детей и подростков развиваются хронические нарушения функций печени и почек
. Болезнь сопровождается накоплением фруктозо-1-ф,
который ингибирует активность фосфоглюкомутазы, поэтому происходит торможение распада гликогена и развивается гипогликемия
. Как следствие, ускоряется мобилизация липидов, окисление жирных кислот и синтез кетоновых тел. Повышение кетоновых тел может привести к метаболическому ацидозу.
Результатом торможения гликогенолиза и гликолиза является снижение синтеза АТФ. Кроме того, накопление фосфорилированной фруктозы ведёт к нарушению обмена неорганического фосфата и гипофосфатемии
. Для пополнения внутриклеточного фосфата ускоряется распад адениловых нуклеотидов. Продукты распада этих нуклеотидов включаются в катаболизм, проходя стадии образования гипоксантина, ксантина и, наконец, мочевой кислоты. Повышение количества мочевой кислоты и снижение экскреции уратов в условиях метаболического ацидоза проявляются в виде гиперурикемии
. Следствием гиперурикемии может быть подагра даже в молодом возрасте.
Метаболизм галактозы
Галактоза образуется в кишечнике в результате гидролиза лактозы. Превращение галактозы в глюкозу происходит в печени в реакции эпимеризации в виде УДФ-производного.
Галактокиназа
(АТФ: галактозо-1-фосфотрансфераза) фосфорилирует галактозу.
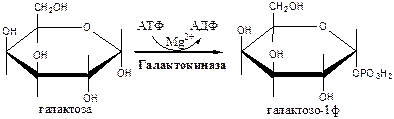
Галактозо-1ф-уридилтрансфераза
замещает галактозой остаток глюкозы в УДФ-глюкозе с образованием УДФ-галактозы.
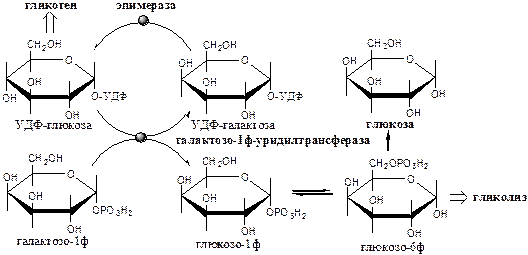
Эпимераза
(УДФ-галактозо-УДФ-глюкозо-изомераза) — НАД-зависимый фермент, катализирует эпимеризацию ОН группы по С4
углеродному атому, обеспечивая взаимопревращения галактозы и глюкозы в составе УДФ.
Образованная глюкозо-1-ф может включаться в: 1) синтез гликогена; 2) превращение в свободную глюкозу; 3) катаболизм, сопряжённый с синтезом АТФ, и т.д.
Нарушения метаболизма галактозы
Галактоземия
обусловленна наследственным дефектом любого из трёх ферментов, включающих галактозу в метаболизм глюкозы.
Галактоземия
, вызванная недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ) имеет несколько форм, проявляется рано, и особенно опасна для детей, так как материнское молоко, содержит лактозу. Ранние симптомы дефекта ГАЛТ: рвота, диарея, дегидратация, уменьшение массы тела, желтуха
. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-ф. В тканях глаза (в хрусталике) галактоза восстанавливается альдоредуктазой (НАДФ) с образованием галактитола (дульцита). Галактитол накапливается в стекловидном теле и связывает большое количество воды, чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения. Галактозо-1-ф ингибирует активность ферментов углеводного обмена (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы).
Галактозо-1ф оказывает токсическое действие на гепатоциты: возникают гепатомегалия, жировая дистрофия. Галактитол и галактозо-1-ф вызывают почечную недостаточность. Отмечают нарушения в клетках полушарий головного мозга и мозжечка, в тяжёлых случаях — отёк мозга, задержку умственного развития, возможен летальный исход.
Некоторые дефекты в строении ГАЛТ приводят лишь к частичной потере активности фермента. Поскольку в норме ГАЛТ присутствует в организме в избытке, то снижение его активности до 50%, а иногда и ниже может клинически не проявляться.
Лечение заключается в удалении галактозы из рациона.
Педфак. Особенности катаболизма моносахаридов у новорожденных и детей
У детей активен УДФ-глюкоза ↔ УДФ-галактоза путь. У взрослых этот путь неактивен. У новорожденных низкая активность ПФШ. При рождении у ребенка происходит переключение катаболизма глюкозы с анаэробного на аэробный путь. Вначале преобладает использование липидов.
ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2005 г
ЛЕКЦИЯ № 9
Тема: Пентозофосфатный шунт и глюконеогенез,
регуляция углеводного обмена.
Факультеты: лечебно-профилактический, медико-профилактический, педиатрический. 2 курс.
Глюконеогенез (ГНГ)
Глюконеогенез
– синтез глюкозы из веществ неуглеводной природы. Его основной функцией является поддержание уровня глюкозы в крови в период длительного голодания и интенсивных физических нагрузок. Основными субстратами глюконеогенеза являются лактат, глицерол, аминокислоты. Глюконеогенез является обратным процессом гликолиза, который протекает в цитоплазме и матриксе митохондрий. Необратимые реакции гликолиза (1, 3 и 10), катализируемые гексокиназами, фруктокиназами и пируваткиназами обходятся с участием 4 специфических ферментов глюконеогенеза: пируваткарбоксилазы, фосфоенолпируват-карбоксикиназы, фруктозо-1,6-фосфотазы и глюкозо-6-фосфотазы. Кроме того, в глюконеогенезе участвуют ферменты ЦТК, например, малат ДГ.
Реакции глюконеогенеза
представлены на схеме. Ключевые (необратимые) реакции глюконеогенеза:
1.
Пируваткарбоксилаза
(ПВК: СО2
-синтетаза (АТФ→АДФ+Фн)) содержит биотин, находиться в митохондриях, превращает ПВК в ЩУК. Индуктор: глюкагон, адреналин, кортизол. Репрессор: инсулин. Ингибитор: АМФ, активатор АцетилКоА. Образующийся ЩУК проходит внутреннюю мембрану митохондрий в своей восстановленной (в виде малата) или аминоформе (в виде аспартата).
2.
Фосфоенолпируваткарбоксикиназа
(ГТФ: ЩУК-2-фосфотрансфераза (декарбоксили-рующая)) находиться в цитоплазме, превращает ЩУК в ФЕП. Индуктор: глюкагон, адреналин, кортизол. Репрессор: инсулин.
3.
Фруктозо-1,6-фосфотаза
(Фруктозо-1,6дф: фосфо-гидролаза) дефосфорилирует фруктозо-1,6дф. Индуктор: глюкагон, адреналин, кортизол. Репрессор: инсулин. Ингибирует АМФ, фруктозо-2,6дф. Активатор: цитрат, жирные кислоты.
4.
Глюкозо-6-фосфотаза
(Глюкозо-6ф: фосфо-гидролаза) дефосфорилирует глюкозо-6ф. Индуктор: глюкагон, адреналин, кортизол. Репрессор: инсулин.
Энергетический баланс глюконеогенеза
. На образование 1 глюкозы из 2 лактатов требуется 6 АТФ: 2 АТФ для пируваткарбоксилазы, 2 ГТФ для ФЕПкарбоксикиназы, 2 АТФ для фосфоглицераткиназы. Обще уравнение глюконеогенеза:
2 лактат + 4 АТФ + 2 ГТФ + 4 Н2
О → 1 глюкоза + 4 АДФ + 2 ГДФ + 6 Фн
Регуляция глюконеогенеза
. Регуляция глюконеогенеза осуществляется реципрокно с реакциями гликолиза: активация глюконеогенеза, сопровождается ингибированием гликолиза и наоборот. Регуляция обмена глюкозы происходит с участием гормонов и метаболитов, которые изменяют активность и количество регуляторных ферментов гликолиза и глюконеогенеза. Инсулин индуцирует синтез ключевых ферментов гликолиза и репрессирует синтез ключевых ферментов глюконеогенеза. Глюкагон, кортизол и адреналин индуцирует синтез ключевых ферментов глюконеогенеза. Ключевые ферменты гликолиза активируют – АМФ, фруктозо-2,6дф, фруктозо-1,6дф, ингибируют – АТФ, НАДН2
, цитрат, жирные кислоты, аланин, АцетилКоА, глюкагон, адреналин. Ключевые ферменты глюконеогенеза активируют – АцетилКоА, глюкагон, ингибируют – АМФ, фруктозо-2,6дф.
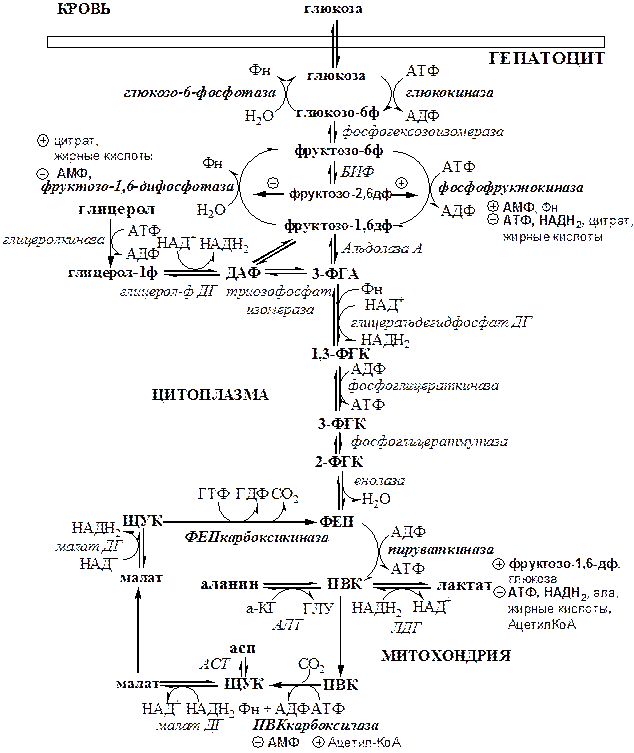 Тканевые особенности глюконеогенеза.
В большинстве тканей глюконеогенеза нет. Тканевые особенности глюконеогенеза.
В большинстве тканей глюконеогенеза нет.
Наибольшая активность глюконеогенеза отмечается в печени, меньше в почках и слизистой оболочке кишечника, в них может синтезироваться до 80-100г глюкозы в сутки. В этих органах глюконеогенез идет до конца с образованием свободной глюкозы, которая может выходить из клеток, поддерживая гомеостаз глюкозы в крови. В норме гомеостаз глюкозы в крови обеспечивается глюконеогенезом печени до 80%, почек до 20%.
Небольшая активность глюконеогенеза наблюдается в мышечных тканях, однако из-за отсутствия у них последних ферментов глюконеогенеза, вместо свободной глюкозы образуются только ее производные, которые не способны покинуть клетку. Таким образом, углеводы синтезируются в мышечных тканях только для собственных нужд. Например, в скелетных мышцах и жировой ткани нет глюкозо-6-фосфотазы, продукт глюконеогенеза – глюкозо-6ф. В миокарде и гладких мышцах нет фруктозо-1,6-дифосфотазы, продукт глюконеогенеза – фруктозо-1,6-дф.
Биологическое значение глюконеогенеза
. Необходимость поддержание постоянного уровня глюкозы в крови связана с тем что, для многих тканей глюкоза является основным (нервная ткань), а для некоторых единственным (эритроциты) источником энергии. Потребность в синтезе глюкозы объясняется тем что, гликогенолиз печени может самостоятельно обеспечивать гомеостаз глюкозы в крови только в течение 8-12 часов, далее запас гликогена в течение суток почти полностью истощается. В условиях длительного голодания (больше суток) глюконеогенез является единственным источником глюкозы в организме.
Пентозофосфатный шунт (ПФШ)
Пентозофосфатный шунт (путь, цикл) является альтернативным путем окисления глюкозы. Наиболее активен этот процесс в жировой ткани, печени, коре надпочечников, эритроцитах, фагоцитирующих лейкоцитах, лактирующей молочной железе, семенниках. Протекает он в цитозоле без участия кислорода и состоит из 2 стадий окислительной и неокислительной. В окислительной стадии происходит восстановление НАДФН2
, который используется: 1) для регенерации глутатиона в антиоксидантной системе; 2) для синтеза жирных кислот; 3) в оксигеназных реакциях с участием цитохрома Р450
при обезвреживании ксенобиотиков, метаболитов, синтезе холестерина, стероидных гормонов и т.д. В неокислительной стадии образуются различные пентозы. Рибозо-5ф может использоваться для синтеза пуриновых и пиримидиновых нуклеотидов.
Тканевые особенности функционирования ПФШ (пути, цикла).
В зависимости от потребности ткани, пентозофосфатный процесс может протекать в виде метаболического цикла, пути или шунта начальных реакций гликолиза:
1.
При ПФЦ или ПФШ в качестве продукта образуется только НАДФН2
. Пентозы в этом случае не являются конечным продуктом, они превращаются в фосфогексозы, которые замыкают цикл, или уходят в гликолиз, завершая шунт. В жировой ткани, эритроцитах.
2.
Продуктом ПФП являются НАДФН2
и пентозы. В печени, костном мозге.
3.
В тканях, которые не испытывают потребность в НАДФН2
, функционирует только неокислительная стадия ПФП, причем ее реакции идут в обратную сторону начиная с фруктозы-6ф до фосфопентоз. В мышцах.
Реакции окислительной стадии
Окислительная стадия ПФШ (пути, цикла) состоит из 3 необратимых реакций:
1). Глюкозо-6ф дегидрогеназа
(глюкозо-6ф: НАДФ+
оксидоредуктаза). Ингибитор НАДФН2
. Индуктор инсулин.
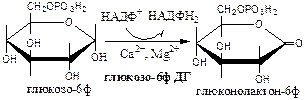
2). Глюконолактонгидратаза
(6-фосфоглюконат: гидро-лиаза).
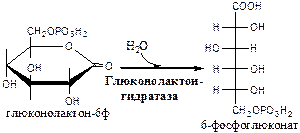
3). 6-фосфоглюконат дегидрогеназа
(6-фосфоглюконат: НАДФ+
оксидоредуктаза (декарбоксилирующая)). Индуктор инсулин.
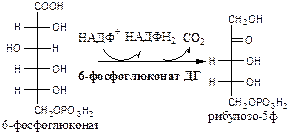
Схема ПФШ (пути, цикла)
На схеме неокислительная стадия начинается с эпимераз и изомераз, которые изомеризуют рибулозо-5ф. Все реакции неокислительной стадии обратимы.
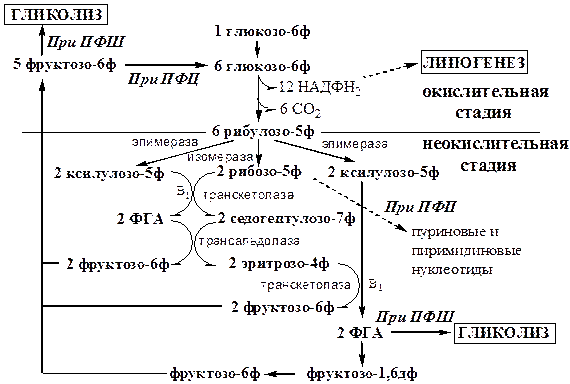
Общее уравнение ПФЦ:
6 глюкозо-6ф + 12 НАДФ+
→ 6 СО2
+ 12 НАДФН2
+ 5 глюкозо-6ф
Общее уравнение ПФШ:
3 глюкозо-6ф + 6 НАДФ+
→ 3 СО2
+ 6 НАДФН2
+ 2 фруктозо-6ф + ФГА
Общие уравнения ПФП:
1) глюкозо-6ф + 2 НАДФ+
→ СО2
+ 2 НАДФН2
+ рибозо-5ф
2) 2 фруктозо-6ф + ФГА → 3 рибозо-5ф
Патология ПФШ
НАДФН2
является важным компонентом антиоксидантной защиты, он необходим для регенерации глутатиона, который с участием глутатионпероксидазы разрушает активные формы кислорода. Так как в эритроцитах НАДФН2
образуется только в реакциях ПФШ, дефект глюкозо-6ф ДГ
вызывает дефицит НАДФН2
и снижение антиоксидантной защиты. В этом случае под действием прооксидантов, например, антималярийных препаратов происходит существенное повышение СРО. Активация СРО вызывает окисление цистеина в белковой части гемоглобина, в результате чего протомеры гемоглобина, соединяясь дисульфидными мостиками, образуют тельца Хайнца. Т.к. тельца Хайнца снижают пластичность клеточной мембраны эритроцитов, она при деформации в капиллярах разрушается. Массированный гемолиз эритроцитов ведет к развитию гемолитической анемии.
Витамин B1
(тиамин).
Структура витамина включает пиримидиновое и тиазоловое кольца, соединённые метановым мостиком.
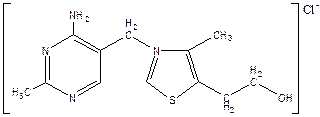
Источники.
Витамин В1
— первый витамин, выделенный в кристаллическом виде К. Фун-ком в 1912 г. Он широко распространён в продуктах растительного происхождения (оболочка семян хлебных злаков и риса, горох, фасоль, соя и др.). В организмах животных витамин В1
, содержится преимущественно в виде дифосфорного эфира тиамина (ТДФ); он образуется в печени, почках, мозге, сердечной мышце путём фосфорилирования тиамина при участии тиаминкиназы и АТФ.
Суточная потребность
взрослого человека в среднем составляет 2-3 мг витамина В1
. Но потребность в нём в очень большой степени зависит от состава и общей калорийности пищи, интенсивности обмена веществ и интенсивности работы. Преобладание углеводов в пище повышает потребность организма в витамине; жиры, наоборот, резко уменьшают эту потребность.
Биологическая роль
витамина В1
, определяется тем, что в виде ТДФ он входит в состав как минимум трёх ферментов и ферментных комплексов: в составе пируват- и α-кетоглутаратдегидрогеназных комплексов он участвует в окислительном декарбокси-лировании пирувата и α-кетоглутарата; в составе транскетолазы ТДФ участвует в пентозофосфатном пути превращения углеводов.
Основной, наиболее характерный и специфический признак недостаточности витамина В1
— полиневрит, в основе которого лежат дегенеративные изменения нервов. Вначале развивается болезненность вдоль нервных стволов, затем — потеря кожной чувствительности и наступает паралич (бери-бери). Второй важнейший признак заболевания — нарушение сердечной деятельности, что выражается в нарушении сердечного ритма, увеличении размеров сердца и в появлении болей в области сердца. К характерным признакам заболевания, связанного с недостаточностью витамина В1
, относят также нарушения секреторной и моторной функций ЖКТ; наблюдают снижение кислотности желудочного сока, потерю аппетита, атонию кишечника.
Регуляция обмена углеводов
Энергетический гомеостаз обеспечивает энергетические потребности тканей с использованием различных субстратов. Т.к. углеводы являются основным источником энергии для многих тканей и единственным для анаэробных, регуляция углеводного обмена является важной составляющей энергетического гомеостаза организма.
Регуляция углеводного обмена осуществляется на 3 уровнях:
1. центральный.
2. межорганный.
3. клеточный (метаболический).
1. Центральный уровень регуляции углеводного обмена
Центральный уровень регуляции осуществляется с участием нейроэндокринной системы и регулирует гомеостаз глюкозы в крови и интенсивность метаболизма углеводов в тканях. К основным гормонам, поддерживающим нормальный уровень глюкозы в крови 3,3-5,5 мМоль/л, относят инсулин и глюкагон. На уровень глюкозы влияют также гормоны адаптации – адреналин, глюкокортикоиды и другие гормоны: тиреоидные, СДГ, АКТГ и т.д.
2. Межорганный уровень регуляции углеводного обмена
Глюкозо-лактатный цикл (цикл Кори) Глюкозо-аланиновый цикл
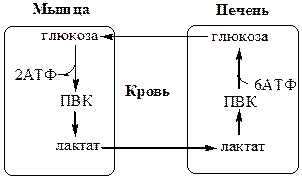 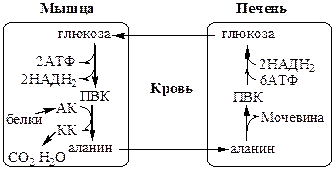
Глюкозо-лактатный цикл
не требует наличие кислорода, функционирует всегда, обеспечивает: 1) утилизацию лактата, образующегося в анаэробных условиях (скелетные мышцы, эритроциты), что предотвращает лактоацидоз; 2) синтез глюкозы (печень).
Глюкозо-аланиновый цикл
функционирует в мышцах при голодании. При дефиците глюкозы, АТФ синтезируется за счет распад белков и катаболизма аминокислот в аэробных условиях, при этом глюкозо-аланиновый цикл обеспечивает: 1) удаление азота из мышц в нетоксичной форме; 2) синтез глюкозы (печень).
3. Клеточный (метаболический) уровень регуляции углеводного обмена
Метаболический уровень регуляции углеводного обмена осуществляется с участием метаболитов и поддерживает гомеостаз углеводов внутри клетки. Избыток субстратов стимулирует их использование, а продукты ингибируют свое образование. Например, избыток глюкозы стимулирует гликогенез, липогенез и синтез аминокислот, дефицит глюкозы - глюконеогенез. Дефицит АТФ стимулирует катаболизм глюкозы, а избыток – наоборот ингибирует.
IV. Педфак
. Возрастные особенности ПФШ и ГНГ, значение.
ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2006 г
ЛЕКЦИЯ № 10
Тема: Структура и обмен инсулина, его рецепторов, транспорт глюкозы.
Механизм действия и метаболические эффекты инсулина.
Факультеты: лечебно-профилактический, медико-профилактический, педиатрический. 2 курс.
Гормоны поджелудочной железы
Поджелудочная железа выполняет в организме две важнейшие функции: экзокринную и эндокринную. Экзокринную функцию выполняет ацинарная часть поджелудочной железы, она синтезирует и секретирует панкреатический сок. Эндокринную функцию выполняют клетки островкового аппарата поджелудочной железы, которые секретируют пептидные гормоны, участвующие в регуляции многих процессов в организме. 1-2 млн. островков Лангерганса составляют 1-2% массы поджелудочной железы.
В островковой части поджелудочной железы выделяют 4 типа клеток, секретирующих разные гормоны: А- (или α-) клетки (25%) секретируют глюкагон, В- (или β-) клетки (70%) — инсулин, D- (или δ-) клетки (<5%) — соматостатин, F-клетки (следовые количества) секретируют панкреатический полипептид. Глюкагон и инсулин в основном влияют на углеводный обмен, соматостатин локально регулирует секрецию инсулина и глюкагона, панкреатический полипептид влияет на секрецию пищеварительных соков. Гормоны поджелудочной железы выделяются в панкреатическую вену, которая впадает в воротную. Это имеет большое значение т.к. печень является главной мишенью глюкагона и инсулина.
Строение инсулина
Инсулин — полипептид, состоящий из двух цепей. Цепь А содержит 21 аминокислотный остаток, цепь В — 30 аминокислотных остатков. В инсулине 3 дисульфидных мостика, 2 соединяют цепь А и В, 1 соединяет 6 и 11 остатки в А цепи.

Инсулин может существовать в форме: мономера, димера и гексамера. Гексамерная структура инсулина стабилизируется ионами цинка, который связывается остатками Гис в положении 10 В-цепи всех 6 субъединиц.
Инсулины некоторых животных имеют значительное сходство по первичной структуре с инсулином человека. Бычий инсулин отличается от инсулина человека на 3 аминокислоты, а инсулин свиньи отличается только на 1 аминокислоту (ала
вместо тре
на С конце В-цепи).
Во многих положениях А и В цепи встречаются замены, не оказывающие влияния на биологическую активность гормона. В положениях дисульфидных связей, остатков гидрофобных аминокислот в С-концевых участках В-цепи и С- и N-концевых остатков А-цепи замены встречаются очень редко, т.к. эти участки обеспечивают формирование активного центра инсулина.
Биосинтез инсулина
включает образование двух неактивных предшественников, препроинсулина и проинсулина, которые в результате последовательного протеолиза превращаются в активный гормон.
1. На рибосомах ЭПР синтезируется препроинсулин (L-В-С-А, 110 аминокислот), биосинтез его начинается с образования гидрофобного сигнального пептида L (24 аминокислот), который направляет растущую цепь в просвет ЭПР.
2. В просвет ЭПР препроинсулин превращается в проинсулин при отщеплении эндопептидазой I сигнального пептида. Цистеины в проинсулине окисляются с образованием 3 дисульфидных мостиков, проинсулин становиться «сложным», имеет 5% активности от инсулина.
3. «Сложный» проинсулин (В-С-А, 86 аминокислот) поступает в аппарат Гольджи, где под действием эндопептидазы II расщепляется с образованием инсулина (В-А, 51 аминокислот) и С-пептида (31 аминокислота).
4. Инсулин и С-пептид включаются в секреторные гранулы, где инсулин соединяется с цинком, образуя димеры и гексамеры. В секреторной грануле содержание инсулина и С-пептида составляет 94%, проинсулина, интермедиатов и цинка - 6%.
5. Зрелые гранулы сливаются с плазматической мембраной, а инсулин и С-пептид попадают во внеклеточную жидкость и далее в кровь. В крови олигомеры инсулина распадаются. За сутки в кровь секретируется 40-50 ед. инсулина, это составляет 20% от его общего запаса в поджелудочной железе. Секреция инсулина энергозависимый процесс, происходит с участием микротубулярно-ворсинчатой системы.

Схема биосинтеза инсулина в β-клетках островков Лангерганса
ЭПР — эндоплазматический ретикулум. 1 — образование сигнального пептида; 2 — синтез препроинсулина; 3 — отщепление сигнального пептида; 4 — транспорт проинсулина в аппарат Гольджи; 5 — превращение проинсулина в инсулин и С-пептид и включение инсулина и С-пептида в секреторные гранулы; 6 — секреция инсулина и С-пептида.
Ген инсулина находиться в 11 хромосоме. Выявлены 3 мутации этого гена, у носителей низкая активность инсулина, отмечается гиперинсулинемия, нет инсулинорезистентности.
Регуляция синтеза и секреции инсулина
Синтез инсулина индуцируют глюкоза и секреция инсулина. Репрессирует секрецию жирные кислоты.
Секрецию инсулина стимулируют: 1. глюкоза
(главный регулятор), аминокислоты (особенно лей и арг); 2. гормоны ЖКТ (β-адренергические агонисты, через цАМФ): ГИП
, секретин, холецистокинин, гастрин, энтероглюкагон; 3. длительно высокие концентрации СТГ, кортизола, эстрогенов, прогестинов, плацентарного лактогена, ТТГ, АКТГ; 4. глюкагон; 5. повышение К+
или Са2+
в крови; 6. лекарства, производные сульфонилмочевины (глибенкламид).
Под влиянием соматостатина секреция инсулина понижается. β-клетки также находятся под влиянием автономной нервной системы. Парасимпатическая часть (холинергические окончания блуждающего нерва) стимулирует выделение инсулина. Симпатическая часть (адреналин через α2
-адренорецепторы) подавляет выделение инсулина.
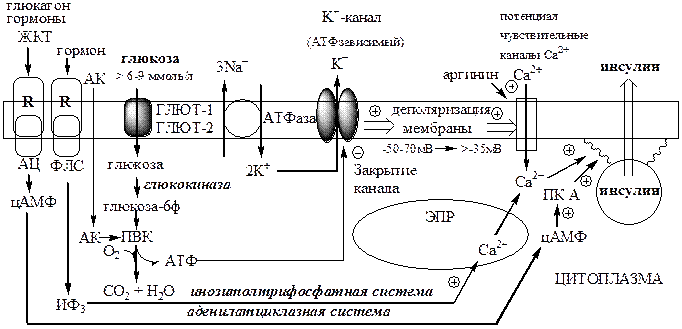
Секреция инсулина осуществляется с участием нескольких систем, в которых основная роль принадлежит Са2+
и цАМФ.
Поступление Са2+
в цитоплазму контролируется несколькими механизмами:
1). При повышении концентрации глюкозы в крови выше 6-9 ммоль/л, она при участии ГЛЮТ-1 и ГЛЮТ-2 поступает в β-клетки и фосфорилируется глюкокиназой. При этом концентрация глюкозо-6ф в клетке прямо пропорциональна концентрации глюкозы в крови. Глюкозо-6ф окисляется с образованием АТФ. АТФ образуется также при окислении аминокислот и жирных кислот. Чем больше в β-клетке глюкозы, аминокислот, жирных кислот тем больше из них образуется АТФ. АТФ ингибирует на мембране АТФ-зависимые калиевые каналы, калий накапливается в цитоплазме и вызывает деполяризацию клеточной мембраны, что стимулирует открытие потенциалзависимых Са2+
-каналов и поступление Са2+
в цитоплазму.
2). Гормоны, активирующие инозитолтрифосфатную систему (ТТГ), выпускают Са2+
из митохондрий и ЭПР.
цАМФ
образуется из АТФ с участием АЦ, которая активируется гормонами ЖКТ, ТТГ, АКТГ, глюкагоном и Са2+
-кальмодулиновым комплексом.
цАМФ и Са2+
стимулируют полимеризацию субъединиц в микротубулы (микроканальцы). Влияние цАМФ на микроканальцевую систему опосредуется через фосфорилирование ПК А микроканальцевых белков. Микроканальцы способны сокращаться и расслабляться, перемещая гранулы по направлению к плазматической мембране обеспечивая экзоцитоз.
Секреция инсулина в ответ на стимуляцию глюкозой представляет собой двухфазную реакцию, состоящую из стадии быстрого, раннего высвобождения инсулина, называемую первой фазой секреции (начинается через 1 мин, продолжается 5-10 мин), и второй фазы (продолжительность ее до 25-30 мин).
Транспорт инсулина.
Инсулин водорастворим и не имеет белка-переносчика в плазме. Т1/2
инсулина в плазме крови составляет 3—10 мин, С-пептида — около 30 мин, проинсулина 20-23 мин.
Разрушение инсулина
происходит под действием инсулинзависимой протеиназы и глутатион-инсулин-трансгидрогеназы в тканях мишенях: в основном в печени (за 1 проход через печень разрушается около 50% инсулина), в меньшей степени в почках и плаценте.
БИОЛОГИЧЕСКИЕ ФУНКЦИИ ИНСУЛИНА
Инсулин
— главный анаболический гормон, он влияет на все виды обмена веществ во всём организме. Однако в первую очередь действие инсулина касается обмена углеводов.
Влияние инсулина на метаболизм глюкозы
Инсулин стимулирует утилизацию глюкозы в клетках разными путями. Около 50% глюкозы используется в процессе гликолиза, 30—40% превращается в жиры и около 10% накапливается в форме гликогена. Общий результат стимуляции этих процессов — снижение концентрации глюкозы в крови.
Влияние инсулина на метаболизм липидов
В печени и жировой ткани инсулин стимулирует синтез липидов, обеспечивая получение для этого процесса необходимых субстратов (ацетил-КоА, глицерофосфат и NADPH2
) из глюкозы. В жировой ткани инсулин тормозит мобилизацию липидов, что снижает концентрацию жирных кислот, циркулирующих в крови.
Влияние инсулина на метаболизм белков
Инсулин оказывает в целом анаболическое действие на белковый обмен. Он стимулирует потребление нейтральных аминокислот в мышцах и синтез белков в печени, мышцах и сердце.
Кроме того, инсулин регулирует
клеточную дифференцировку, пролиферацию и трансформацию большого количества клеток. Инсулин поддерживает рост и репликацию многих клеток эпителиального происхождения, в том числе гепатоцитов, опухолевых клеток. Инсулин усиливает способность фактора роста фибробластов (ФРФ), тромбоцитарного фактора роста (ТФР), фактора роста эпидермиса (ФРЭ), простагландина (ПГF2
a), вазопрессина и аналогов цАМФ активировать размножение клеток.
Основные направления действия инсулина
1. Инсулин регулирует транспорт веществ
Инсулин стимулирует транспорт в клетку глюкозы, аминокислот, нуклеозидов, органического фосфата, ионов К+
и Са2+
. Эффект проявляются очень быстро, в течение нескольких секунд и минут.
Транспорт глюкозы в клетки происходит при участии ГЛЮТ. В мышцах и жировой ткани инсулинзависимый ГЛЮТ-4, в отсутствие инсулина находится в цитозольных везикулах. Под влиянием инсулина происходит транслокация везикул с ГЛЮТ в плазматическую мембрану и начинается транспорт глюкозы. При снижении концентрации инсулина, ГЛЮТ-4 возвращаются в цитозоль, и транспорт глюкозы прекращается.
2. Инсулин регулирует синтез ферментов
Инсулин влияет на скорость транскрипции более чем 100 специфических мРНК в печени, жировой ткани, скелетных мышцах и сердце. Эффект реализуется в течение несколько часов. В клетках печени инсулин индуцирует синтез ключевых ферментов гликолиза (глюкокиназы, фруктокиназы и пируваткиназы), ПФШ (глюкозо-6ф ДГ), липогенеза (цитратлиаза, пальмитатсинтаза, Ацетил-КоА-карбоксилаза), транспортеров глюкозы (?) и репрессирует синтез ключевого фермента глюконеогенеза (ФЕП карбоксикиназу).
3. Инсулин регулирует активность ферментов
Инсулин регулирует активность ферментов путем их фосфорилирования и дефосфорилирования. Эффект проявляются очень быстро, в течение нескольких секунд и минут.
· Инсулин активирует ключевые ферменты гликолиза: в печени, мышцах, жировой ткани – фосфофруктокиназу и пирруваткиназу; в печени – глюкокиназу; в мышцах - гексокиназу II.
· Инсулин ингибирует в печени глюкозо-6-фосфотазу, что тормозит глюконеогенез и выход глюкозы в кровь.
· Инсулин активирует фосфопротеинфосфотазу гликогенсинтазы и гликогенфосфорилазы, в результате активируется синтез гликогена и тормозится его распад.
· В адипоцитах инсулин активирует ключевой фермент липогенеза (АцетилКоА-карбоксилазу). Инсулин в гепатоцитах и адипоцитах активирует фосфопротеинфосфатазу, которая дефосфорилирует и инактивирует ТАГ-липазу, что тормозит липолиз.
· Инсулин снижает активность аминотрансфераз и ферментов цикла мочевины. Последний эффект инсулина характеризуется повышением активности РНК-полимеразы и концентрации РНК в печени. При этом увеличивается скорость образования полисом и рибосом.
· Инсулин активирует ФДЭ, которая снижает концентрацию цАМФ, прерывает эффекты контринсулярных гормонов: в печени и жировой ткани тормозит липолиз, в печени и мышцах - глюконеогенез.
МЕХАНИЗМ ДЕЙСТВИЯ ИНСУЛИНА
Инсулин связывается с инсулиновым рецептором (IR), находящимся на мембране. IR обнаружены почти во всех типах клеток, но больше всего их в гепатоцитах и клетках жировой ткани (концентрация достигает до 20000 на клетку). IR постоянно синтезируется (ген в 19 хромосоме) и разрушается. После связывания инсулина с IR весь комплекс погружается в цитоплазму, достигает лизосом, где инсулин разрушается, а IR может разрушаться, а может возвращаться мембрану. Т1/2
IR 7—12 ч, но в присутствии инсулина уменьшается до 2-3 ч.
При высокой концентрации инсулина в плазме крови, число IR может уменьшаться в результате усиленного разрушения в лизосомах. Также у IR может снижаться активность при его фосфорилировании по остаткам серина и треонина.
Рецептор инсулина
(
IR)
- гликопротеин, состоит из 2 α и 2 β субъединиц связанных дисульфидными связями. α субъединицы (719 АК) расположены вне клетки, они связывают инсулин, а β субъединицы (трансмебранный белок, 620 АК) обладают тирозинкиназной активностью. После присоединения гормона к α субъединицам, β субъединицы сначала фосфорилируют друг друга, а затем внутриклеточные белки — субстраты инсулинового рецептора (IRS). Известно несколько таких субстратов: IRS-1, IRS-2 (фосфопротеины, состоящие из более чем 1200 аминокислот), Shc, а также некоторые белки семейства STAT.
Активация инсулином сигнального пути
Ras
Фосфорилированный инсулиновым рецептором She соединяется с небольшим цитозольным белком Grb. К образовавшемуся комплексу присоединяется с Ras-белок (из семейства малых ГТФ-связывающих белков, в неактивном состоянии прикреплён к внутренней поверхности плазматической мембраны и связан с ГДФ), GAP (от англ. GTP-
ase
activating
factor —
фактор, активирующий ГТФазу), GEF (от англ. GTP
exchange
factor
— фактор обмена ГТФ) и SOS (от англ. son
ofsevenless,
названный по мутации гена у дрозофилы). Два последних белка способствуют отделению ГДФ от Ras-белка и присоединению к нему ГТФ, с образованием активной ГТФ-связанной формы Ras.
Активированный Ras соединяется с протеинкиназой Raf-1 и активирует ее в результате многоэтапного процесса. Активированная ПК Raf-1 стимулирует каскад реакций фосфорилирования и активации других протеинкиназ. ПК Raf-1 фосфорилирует и активирует киназу МАПК, которая, в свою очередь, фосфорилирует и активирует митогенактивируемые протеинкиназы МАПК.
МАПК фосфорилирует многие цитоплазматические белки: ПК pp90S6, белки рибосом, фосфолипазу А2
, активаторы транскрипции STAT.
В результате активации протеинкиназ происходит фосфорилирование ферментов и факторов транскрипции, что составляет основу многочисленных эффектов инсулина. Например:
Активация гликогенсинтазы
ПК pp90S6 фосфорилирует и активирует фосфопротеинфосфатазу (ФПФ). ФПФ дефосфорилирует и инактивирует киназу гликогенфосфорилазы и гликогенфосфорилазу, дефосфорилирует и активирует гликогенсинтазу. В результате активируется синтез гликогена, а распад - ингибируется.
Активация инозитолтрифосфатной системы
Фосфорилированные инсулином белки IRS-1 присоединяются к ФЛ С и активируют ее.
ФЛ С расщепляет фосфатидилинозитолы с образованием инозитолфосфатов и ДАГ.
Фосфорилированные инсулином белки IRS-1 и Shc присоединяются к фосфоинозитол-3-киназе (ФИ-3-киназа) и активируют ее.
ФИ-3-киназа катализирует фосфорилирование инозитолфосфатов (ФИ, ФИ-4-ф и ФИ-4,5-бф) в 3 положении, образуя инозитолполифосфаты: ФИ-3-ф, ФИ-3,4-бф, ФИ-3,4,5-тф. ФИ-3,4,5-тф (ИФ3
) стимулирует мобилизацию Са2+
из ЭПР.
Са2+
и ДАГ активирует специфические ПК С.
Са2+
активирует микроканальцы, которые осуществляют транслокацию ГЛЮТ-4 в плазматическую мембрану, и таким образом ускоряет трансмембранный перенос глюкозы в клетки жировой и мышечной ткани.
Активация фосфодиэстеразы
Фосфорилированные инсулином белки IRS-1 и Shc присоединяются к протеинкиназе В (ПК В) и активируют ее. ПК В фосфорилирует и активирует фосфодиэстеразу (ФДЭ). ФДЭ катализирует превращение цАМФ в АМФ, прерывая эффекты контринсулярных гормонов, что приводит к торможению липолиза в жировой ткани, гликогенолиза в печени.
Регуляция транскрипции мРНК
STAT – особые белки, являются переносчиками сигнала и активаторами транскрипции. При фосфорилировании STAT с участием IR или МАПК образуют димеры, которые транспортируются в ядро, где связываются со специфическими участками ДНК, регулируют транскрипцию мРНК и биосинтез белков-фементов.
Путь Ras активируется не только инсулином, но и другими гормонами и факторами роста.
ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2006 г
ЛЕКЦИЯ № 11
Тема: Сахарный диабет I и II типа: механизмы возникновения,
метаболические нарушения, осложнения.
Факультеты: лечебно-профилактический, медико-профилактический, педиатрический.
2 курс.
В норме уровень глюкозы в крови натощак составляет 3.3 – 5.5 ммоль/л.
Гипергликемия
– повышение уровня глюкозы в крови выше 6,1 ммоль/л. Гипергликемия бывает физиологической и патологической.
Причины физиологической гипергликемии:
1) алиментарная,
при употреблении легкоусвояемых углеводов. Не превышает 11 ммоль/л, нормализуется в течение 3 часов;
2) стрессорная
, под действием катехоламинов, глюкокортикоидов, вазопрессина;
3) кратковременные физические нагрузки
.
Причины патологической гипергликемии:
1) судороги
при эпилепсиях, столбняке;
2) эндокринные нарушения
. Гиперпродукция контринсулярных гормонов (гипертириоз, синдромы Кушинга и Кона), абсолютный или относительный дефицит инсулина (сахарный диабет).
3) ЧМТ
.
Гипогликемия
снижение уровня глюкозы в крови ниже 3,3 ммоль/л. Гипогликемия бывает физиологической и патологической.
Причины физиологической гипогликемии: 1) алиментарная,
при голодании; 2) длительная физическая нагрузка
.
Причины патологической гипогликемии: 1) эндокринные нарушения
при избытке инсулина (инсулинома – доброкачественная опухоль β-клеток, передозировка инсулина у больных СД) или недостаточности контринсулярных гормонов (гипотиреоз, дефицит глюкокортикоидов); 2) гликогенозы, агликогенозы,
препятствующие гликогенолизу; 3) печеночная недостаточность, связанная с низкой активностью глюконеогенеза; 4) почечная недостаточность, связанная с врожденной патологией реабсорбции глюкозы (почечный диабет); 5) отравления монойодацетатом (вызывает глюкозурию).
Сахарный диабет
(СД) — системное гетерогенное заболевание, обусловленное абсолютным или относительным дефицитом инулина, который сначала вызывает нарушение углеводного, а затем всех видов обмена, что в итоге поражает все функциональные системы организма.
СД широко распространенное заболевание, им страдает 6,6% населения, в России – 5%.
СД бывает первичным и вторичным. Кроме того, выделяют нарушение толерантности к глюкозе и СД беременных.
Первичный СД
- самостоятельное заболевание.
Вторичный СД
является симптоматическим, он возникает при патологии эндокринных желез (акромегалия, феохромоцитома, глюкагонома, синдромы Кушинга, Кона) и патологии поджелудочной железы (хронический панкреатит, рак, панкреатэктомия, гемохроматоз, генетические синдромы).
Первичный СД по механизму развития подразделяется на СД I типа (раньше ИЗСД) и СД II типа (раньше ИНСД).
Общими симптомами любого СД являются жажда, полиурия, кожный зуд, склонность к инфекциям.
Этиологическая классификация СД (ВОЗ 1999)
.
1. Сахарный диабет
I типа (раньше ИЗСД)
а). Аутоиммунный
б). Идиопатический
2. СД
II типа (раньше ИНСД)
3. Другие специфические типы
а). генетические дефекты β-клеток
б). генетические дефекты в действии инсулина
в). болезни экзокринной части поджелудочной железы (панкреатит и т.д.)
г). эндокринопатии
д). СД, индуцированный лекарствами и химикатами (глюкокортикоиды, никотиновая кислота, тиреоидные гормоны, тиазиды, вакор, пентамидин и т.д.)
е). Инфекции (врожденная краснуха, цитомегаловирус и т.д.).
ж). Необычные формы иммуноопосредованного диабета.
з). Другие генетические синдромы, иногда сочетающиеся с диабетом (Дауна, Тернера и т.д.).
4. Гестационный СД (беременных)
САХАРНЫЙ ДИАБЕТ
I типа
СД
I типа
— заболевание, которое возникает вследствие абсолютного дефицита инсулина, вызванного аутоиммунным разрушением β-клеток поджелудочной железы. СД I типа поражает в большинстве случаев детей, подростков и молодых людей до 30 лет, но может проявиться в любом возрасте. СД I типа редко является семейным заболеванием (10-15% всех случаев).
Причины СД
I типа
1. Генетическая предрасположенность
. Генетические дефекты ведущие к СД могут реализоваться в клетках иммунной системы и β-клетках поджелудочной железы. В β-клетках известно около 20 генов, способствующих развитию СД I типа. В 60-70% случаях СД I типа связан с наличием в 6 хромосоме HLA региона генов DR3, DR4 и DQ.
2. Действие на β-клетки β-цитотропных вирусов
(оспа, краснуха, корь, паротит, Коксаки, аденовирус, цитомегаловирус), химических и других диабетогенов
.
Вариант 1
При наличии генетического дефекта, на поверхности β-клеток накапливаются антигены, имеющие схожую аминокислотную последовательность с β-цитотропными вирусами.
В случае возникновения инфекции β-цитотропных вирусов, развиваются иммунные реакции против этих вирусов и аутоиммунные реакции против схожих антигенов β-клеток. Реакция идет с участием моноцитов, Т-лимфоцитов, антител к β-клеткам, инсулину, глутамат декарбоксилазе (фермент 64кДа, находиться на мембране β-клеток). В результате аутоиммунные реакции вызывают гибель β-клеток.
Вариант 2
При действии на β-клетки с генотипом HLA β-цитотропных вирусов или диабетогенов на поверхности β-клеток происходит изменение антигенов.
На измененные антигены β-клетки развиваются аутоиммунные реакции. Аутоиммунные реакции вызывают гибель β-клеток.
Вариант 3
β-цитотропные вирусы имеют схожую последовательность аминокислот с глутамат декарбоксилазой β-клеток. Генетический дефект СД8+ лимфоцитов (Т-супрессоров) не позволяет им отличить аминокислотную последовательность вируса и глутамат декарбоксилазы,
поэтому при возникновении инфекции, Т-лимфоциты реагируют на глутамат декарбоксилазу β-клеток как на вирус.
Вариант 4
Некоторые β-цитотропные вирусы и химические диабетогены, например, производные нитрозомочевины, нитрозамины, аллоксан самостоятельно и избирательно поражают β-клетки, вызывая их лизис;
Стадии развития СД
I типа
1. Стадия генетической предрасположенности
. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
2. Стадия провоцирующих событий
. Инфекция β-цитотропных вирусов или действие химических диабетогенов. Протекает без клинических симптомов;
3. Стадия явных иммунных аномалий
. Развитие смешанных аутоиммунных реакций против β-клеток. Ресурсы инсулина достаточны. Протекает без клинических симптомов. Развивается от 2-3 месяцев до 2-3 лет;
4. Стадия латентного диабета
. Гибель 75% β-клеток, небольшое снижение инсулина, гипергликемия при нагрузочных пробах, снижение аутоиммунных процессов. Протекает без клинических симптомов;
5. Явный диабет
. Гибель 80-90% β-клеток, заметное снижение инсулина, гипергликемия натощак, нет или слабые аутоиммунные реакции. Появляются клинические симптомы. Развивается 2 года. Необходима инсулинотерапия;
6. Терминальный диабет
. Полная гибель β-клеток, высокая потребность в инсулинотерапии, аутоиммунные проявления снижены или их нет. Выраженные клинические проявления, появляются ангиопатии. Развивается до 3,5 лет;
Изменения метаболизма при СД
I типа
При СД I типа исчезает инсулин, т.к. инсулин ингибитор секреции глюкагона, в крови происходит увеличение глюкагона.
Изменения в углеводном обмене
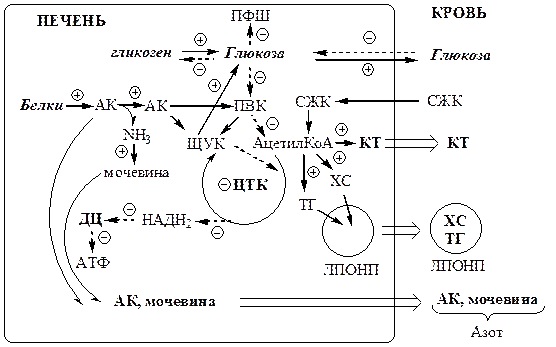
В печени дефицит инсулина и избыток глюкагона стимулирует реакции глюконеогенеза, гликогенолиза и ингибирует реакции гликолиза, ПФШ и синтеза гликогена. В результате в печени глюкозы больше образуется, чем потребляется.
Так как реакции глюконеогенеза протекают через ЩУК, он, образовавшись из ПВК, аспартата и малата, активно вовлекается в глюконеогенез, вместо того чтобы включаться в ЦТК. В результате ЦТК и ДЦ тормозится, снижается образование АТФ, возникает энергодефицит
.
В инсулинзависимых тканях (мышцы, жировая ткань) дефицит инсулина препятствует поступлению глюкозы в клетки и ее использованию в реакциях гликолиза, ПФШ и синтеза гликогена. Блокирование ЦТК и ДЦ также вызывает энергодефицит.
Снижение потребления глюкозы инсулинзависимыми тканями и усиление ее образования в печени приводит к гипергликемии
. Когда гипергликемия превышает концентрационный почечный порог возникает глюкозурия.
Глюкозурия
– наличие глюкозы моче. В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу. Если уровень глюкозы превышает в крови 9-10 ммоль/л, глюкоза не успевает полностью реабсорбироваться из первичной мочи и частично выводится с вторичной мочой.
У больных с СД после приёма пищи концентрация глюкозы в крови может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность
к глюкозе.

Изменения в липидном обмене
Дефицит АТФ, НАДФН2
, инсулина и избыток глюкагона тормозят липогенез и усиливают липолиз в жировой ткани. В результате в крови повышается концентрация свободных жирных кислот, которые поступают в печень и окисляются там до Ацетил-КоА. АцетилКоА при дефиците ЩУК не может включаться в ЦТК. Поэтому он накапливается и поступает на альтернативные пути: синтез кетоновых тел (ацетоуксусная, β-гидроксимасляная кислоты) и холестерина.
В норме кетоновые тела являются источником энергии для аэробных тканей, они превращаются в АцетилКоА, который окисляется в ЦТК. Так как ЦТК заблокирован дефицитом ЩУК, кетоновые тела накапливаются в крови и вызывают кетонемию
. Кетонемия усугубляет недостаточность инсулина, подавляя остаточную секреторную активность β-клеток. Когда кетонемия превышает концентрационный почечный порог (выше 20 мг/дл, иногда до 100 мг/дл) возникает кетонурия. Кетонурия
– наличие кетоновых тел в моче.
В тканях ацетоуксусная кислота частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии.
Липопротеины крови поставляют субстраты для липогенеза в тканях. Дефицит инсулина блокирует липогенез в жировой ткани, ингибирует липопротеинлипазу в сосудах, это препятствует расщеплению липопротеинов крови (в основном, ЛПОНП), в результате они накапливаются, вызывая гиперлипопротеинемию.
Изменения в белковом обмене
Энергодефицит, недостаток инсулина и избыток глюкагона приводит к снижению скорости синтеза белков в организме и усилению их распада, что повышает концентрацию аминокислот в крови. Аминокислоты поступают в печень и дезаминируются до кетокислот. Кетокислоты включаются в глюконеогенез, что усиливает гипергликемию. Из аммиака активно синтезируется мочевина. Повышение в крови аммиака, мочевины, аминокислот вызывает азотемию
– увеличение концентрации азота в крови. Азотемия приводит к азотурии
– увеличению концентрации азота в моче. Развивается отрицательный азотистый баланс. Катаболизм белков ведет к миодистрофии и вторичному иммунодефициту.
Изменения в водно-солевом обмене
Поскольку возможности почек ограничены, высокие концентрации глюкозы, кетоновых тел и мочевины не успевают реабсорбироваться из первичной мочи. Они создают в первичной моче высокое осмотическое давление, которое препятствует реабсорбции воды в кровь и образованию вторичной мочи. У таких пациентов развивается полиурия
, выделение мочи возрастает до 3—4 л в сутки (в некоторых случаях до 8—9 л). Потеря воды вызывает постоянную жажду или полидипсию
. Без частого питья, полиурия может приводить к обезвожива
нию
организма. Потеря с мочой глюкозы усугубляет энергодефицит, может увеличить аппетит и полифагию
. С первичной мочой из организма уходят некоторые полезные минеральные компоненты, что приводит к нарушению минерального обмена.
Высокие концентрации глюкозы, кетоновых тел и мочевины создают в плазме крови значительное осмотическое давление, которое способствует дегидратации
тканей. Кроме воды ткани теряют электролиты, прежде всего ионы К+
, Na+
, С1-
, НСО3
-
.
Изменение в газообмене тканей
Общая дегидратация организма, вызванная полиурией и дегидратацией тканей приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Причиной гипоксии является также гликозилирование Hb в HbA
1
c
, который не переносит О2
к тканям. Гипоксия ведет к энергодефициту и накоплению в организме лактата
.
Изменения в кислотно-основном равновесии
Накопление кетоновых тел, лактата и потеря щелочных валентностей с мочой снижает буферную ёмкость крови и вызывает ацидоз
.
Симптомы СД I типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены. Общая слабость, похудание, снижение трудоспособности, сонливость. Ожирение отсутствует. Повышенный аппетит при кетоацидозе сменяется анорексией. Развивается быстро, склонен к развитию кетоацидотической комы.
САХАРНЫЙ ДИАБЕТ
II типа
СД II типа представляет собой группу гетерогенных нарушений углеводного обмена. СД II типа не инсулинозависимый, не склонен к кетоацидотической коме, не имеет антител к β-клеткам, не аутоиммунной природы, не имеет связи с определенными HLA фенотипами. Ожирение в 80%. На долю СД II типа приходится примерно 85-90% всех случаев СД, он поражает людей, как правило, старше 40 лет и характеризуется высокой частотой семейных форм (риск СД II типа у ближайших родственников больного достигает 50%, тогда как при СД I типа он не превышает 10%). СД II типа поражает преимущественно жителей развитых стран, особенно горожан.
В основе СД II типа лежат множество причин. СД II типа развивается при:
· генетических дефектах рецепторов инсулина, у них снижается чувствительность к инсулину;
· синтезе дефектного инсулина с низкой биологической активностью (мутация гена инсулина: в позиции 24 В-цепи вместо фен присутствует лей);
· нарушении превращения проинсулина в инсулин;
· нарушении секреции инсулина;
· повреждении инсулина и его рецепторов антителами;
· повышения скорости катаболизма инсулина;
· действия контринсулярных гормонов (создают гипеинсулинемию, которая вызывает инсулинорезистентность);
· нарушении глюкозочувствительного механизма b-клеток (мутации гена глюкокиназы) и т.д.
Основным провоцирующим фактором СД II типа служит ожирение.
Стадии СД II типа
1. Стадия генетической предрасположенности
. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
2. Стадия латентного диабета
. Гипергликемия при нагрузочных пробах. Протекает без клинических симптомов СД;
3. Явный диабет
. Гипергликемия натощак. Появляются клинические симптомы.
Симптомы СД II типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены умеренно или отсутствуют. Часто ожирение (у 80-90% больных).
Изменения метаболизма при СД II типа
Относительный дефицит инсулина вызывает метаболические нарушения, схожие с теми которые возникают при абсолютном дефиците инсулина, однако эти нарушения менее выражены, а у 50% больных с ожирением и умеренной гипергликемией СД II типа вообще протекает бессимптомно.
В отличие от абсолютного дефицита инсулина, при относительном дефиците инсулина, влияние инсулина сохраняется на жировую ткань, имеющую высокое содержание рецепторов к инсулину. Инсулин в жировой ткани стимулирует липогенез, блокирует липолиз и выход жирных кислот в кровь, поэтому при СД II типа не наблюдается кетоацидоз, масса тела не уменьшается, а наоборот развивается ожирение. Таким образом, ожирение, с одной стороны, важнейший фактор риска, а с другой — одно из ранних проявлений СД II типа.
Так как синтез инсулина как правило не нарушен, высокий уровень глюкозы в крови стимулирует секрецию инсулина из β-клеток, вызывая гиперинсулинемию
. Высокая концентрация инсулина вызывает инактивацию и разрушение инсулиновых рецепторов, что снижает толерантность тканей к глюкозе. Инсулин больше не может нормализовать гликемию, возникает инсулинорезистентность
. При этом, высокий уровень глюкозы в крови снижает чувствительность β-клеток к глюкозе, в результате запаздывает или отсутствует первая фаза секреции инсулина.
При СД II типа наблюдается гиперинсулинемия (80%), артериальная гипертензия (50%), гиперлипидемия (50%), атеросклероз, нейропатия (15%) и диабетическая нефропатия (5%).
Осложнения СД
Острые осложнения сахарного диабета. Механизмы развития диабетической комы
Острые осложнения специфичны для СД I и II типа.
Дегидратация тканей головного мозга в первую очередь, а также нарушения обмена веществ в нервной ткани могут приводить к развитию острых осложнений в виде коматозных состояний. Кома
это крайне тяжелое состояние, характеризующееся глубоким угнетением ЦНС, стойкой потерей сознания, утратой реакций на внешние раздражители любой интенсивности. Коматозные состояния при СД могут проявляться в трёх формах: кетоацидотической, гиперосмолярной и лактоацидотической.
Кетоацидотическая кома
возникает при СД I типа, когда концентрация кетоновых тел становится выше 100 мг/дл (до 400-500мг/дл).
Гиперкетонемия приводит к:
1) ацидозу, который блокирует активность большинства ферментов, в первую дыхательных, что вызывает гипоксию и снижение синтеза АТФ.
2) гиперосмолярности, которая приводит к дегидратации тканей и нарушению водно-электролитного равновесия, с потерей ионов калия, натрия, фосфора, магния, кальция, бикарбонатов.
Это при определенной выраженности и вызывает коматозное состояние с падением артериального давления и развитием острой почечной недостаточности.
Возникающая гипокалиемия ведет к гипотонии гладкой и поперечно-полосатой мускулатуры, снижению тонуса сосудов, падению АД, сердечной аритмии, гипотонии дыхательной мускулатуры с развитием острой дыхательной недостаточности; атонии ЖКТ с парезом желудка и развитием кишечной непроходимости развивается выраженная гипоксия. В общей причине смертности она занимает 2-4 %.
Гиперосмолярная кома
характерна
для СД II типа, она наблюдается при высокой гипергликемии. У большинства высокая гипергликемия обусловлена сопутствующим нарушением функции почек, ее провоцируют стресс, травма, резкая дегидратация организма (рвота, диарея, ожоги, кровопотеря и т.д.). Гиперосмолярная кома развивается медленно, в течение нескольких дней при беспомощности человека (некомпенсируемая питьем), когда содержание глюкозы достигает 30-50 ммоль/л.
Гипергликемия
способствует полиурии, создает гиперосмотическое состояние
, которое вызывает дегидратацию
тканей, приводящую к нарушению водно-электролитного равновесия.
Резкая дегидротация организма рвотой, диарей, кровопотерей на фоне полиурии и отсутствия питья приводит к гиповолемии
. Гиповолемия
вызывает снижение АД, сгущение крови, увеличение ее вязкости и способности к тромбообразованию
. Нарушение гемодинамики приводит к ишемии
тканей, развитию гипоксии, накоплению лактата и энергодефициту. Ишемия почек приводит к развитию острой почечной недостаточности – анурии
. Анурия приводит к накоплению в крови остаточного азота (аммиак, мочевина, аминокислоты), возникает гиперазотемия
. Гиповолемия через альдостерон снижает выведение с мочой NaCl, что вызывает гипернатриемию и гиперхлоремию
. Гиперазотемия, гипернатриемия и гиперхлоремия усиливают гиперосмотическое состояние и нарушение водно-электролитного равновесия.
Энергодефицит и нарушение водно-электролитного равновесия препятствует формированию на мембране нейронов потенциала и проведению нервных импульсов в ЦНС, что приводит к развитию комы. Смертность при гипергликемической коме 50%.
Лактоацидотическая кома
характерна
для СД II типа, она возникает при накоплении лактата.
В присутствии молочной кислоты резко снижается чувствительность адренорецепторов к катехоламинам, развивается необратимый шок. Появляется метаболическая коагулопатия, проявляющаяся ДВС-синдромом, периферическими тромбозами, тромбоэмболиями (инфаркт миокарда, инсульт).
Ацидоз при избытке кетоновых тел и лактата затрудняет отдачу Hb кислорода в ткани (гипоксия), он блокирует активность большинства ферментов, в первую очередь подавляется синтез АТФ, активный транспорт и создание мембранных градиентов, что в нервной ткани угнетает проведение нервных импульсов и вызывает кому.
Поздние осложнения сахарного диабета
Поздние осложнения СД неспецифичны (возникают при разных видах СД), к ним относятся:
1. макроангиопатия (атеросклероз крупных артерий);
2. нефропатия;
3. ретинопатия;
4. нейропатия;
5. синдром диабетической стопы.
Главная причина поздних осложнений сахарного диабета является гипергликемия, гиперлипидемия и гиперхолестеринемия. Они приводят к повреждению кровеносных сосудов и нарушению функций различных органов и тканей путем гликозилирования белков, образования сорбитола и активации атеросклероза.
1. Неферментативное
гликози
лирование
белков
.
Глюкоза взаимодействует со свободными аминогруппами белков с образованием Шиффовых оснований, при этом белки изменяют свою конформацию и функции. Степень гликозилирования белков зависит от скорости их обновления и концентрации глюкозы.
При гликозилировании кристаллинов - белков хрусталика, образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта
.
При гликозилировании белков (протеогликаны, коллагены, гликопротеины) базальных мембран нарушается их обмен, соотношение и структурная организация, происходит утолщение базальных мембран и развитие ангиопатий
.
Макроангиопатии
проявляются в поражениях крупных и средних сосудов сердца, мозга, нижних конечностей. Гликозилированные белки базальных мембран и межклеточного матрикса (коллагена и эластина) снижают эластичности артерий. Гликозилирование в сочетании с гиперлипидемией гликозилированных ЛП и гиперхолестеринемией является причиной активации атеросклероза.
Микроангиопатии
— результат повреждения капилляров и мелких сосудов. Проявляются в форме нефро-, нейро- и ретинопатии.
Нефропатия
развивается примерно у трети больных СД. Признаком ранних стадий нефропатии служит микроальбуминурия (в пределах 30—300 мг/сут), которая в дальнейшем развивается до классического нефротического синдрома, характеризующегося высокой протеинурией, гипоальбуминемией и отёками.
Ретинопатия,
самое серьёзное осложнение сахарного диабета и наиболее частая причина слепоты, развивается у 60-80% больных СД. На ранних стадиях развивается базальная ретинопатия, которая проявляется в кровоизлияниях в сетчатку, расширении сосудов сетчатки, отёках. Если изменения не затрагивают жёлтого пятна, потеря зрения обычно не происходит. В дальнейшем может развиться пролиферативная ретинопатия, проявляющаяся в новообразовании сосудов сетчатки и стекловидного тела. Ломкость и высокая проницаемость новообразованных сосудов определяют частые кровоизлияния в сетчатку или стекловидное тело. На месте тромбов развивается фиброз, приводящий к отслойке сетчатки и потере зрения.
2. Превращение глюкозы в сорбитол
.
При гипергликемии этот процесс ускоряется. Реакция катализируется альдозоредуктазой. Сорбитол не используется в клетке, а скорость его диффузии из клеток невелика. При гипергликемии сорбитол накапливается в сетчатке и хрусталике глаза, клетках клубочков почек, шванновских клетках, в эндотелии. Сорбитол в высоких концентрациях токсичен для клеток, он приводит к увеличению осмотического давления, набуханию клеток и отёку тканей. При накоплении сорбитола в хрусталике приводит к набуханию и нарушению упорядоченной структуры кристаллинов, в результате хрусталик мутнеет.
Диагностика сахарного диабета
Диагноз сахарного диабета ставят на основе классических симптомов сахарного диабета — полиурии, полидипсии, полифагии, ощущения сухости во рту.
Биохимическими признаками СД являются:
• Уровень глюкозы натощак в капиллярной крови выше 6,1 ммоль/л;
• Уровень С-пептида натощак менее 0,4 ммоль/л – признак СД I типа.
• Тест с глюкагоном. Натощак определяется концентрация С-пептида (в норме >0,6 ммоль/л), затем 1мг глюкагона вводят внутривенно, через 6 минут определяется концентрация С-пептида (в норме >1,1 ммоль/л).
• Наличие глюкозурии (определяют для контроля лечения);
• Глюкозотелерантный тест (ГТТ), проводится при отсутствии клинических симптомов СД, когда концентрация глюкозы в крови натощак соответствует норме. Признак СД - уровень глюкозы в плазме крови выше 11,1 ммоль/л через 2 ч после сахарной нагрузки;
| 
|
Определение толерантности к глюкозе
Обследуемый принимает раствор глюкозы (250-300 мл воды + глюкоза 1 г на 1 кг массы тела). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 — у здорового человека; 2 — у больного сахарным диабетом.
|
Для оценки компенсации СД определяют:
• В норме уровень гликозилированного гемоглобина НbА1с
не более 6% от общего содержания Hb, при компенсированном СД НbА1с
< 8,5%;
• альбуминурии. В норме альбуминов в моче < 30 мг/сут. При сахарном диабете до 300 мг/сут.
Поскольку СД II типа развивается значительно медленнее, классические клинические симптомы, гипергликемию и дефицит инсулина диагностируют позднее, часто в сочетании с симптомами поздних осложнений сахарного диабета.
Лечение сахарного диабета
Лечение сахарного диабета зависит от его типа (I или II), является комплексным и включает диету, применение сахаропонижающих средств, инсулинотерапию, а также профилактику и лечение осложнений.
Сахаропонижающие препараты делят на две основные группы: производные сульфонилмочевины и бигуаниды.
Препараты сульфонилмочевины
блокируют АТФ-чувствительные К+
-каналы, что повышает внутриклеточную концентрацию К+
и приводит к деполяризации мембраны. Деполяризация мембраны ускоряет транспорт ионов кальция в клетку, вследствие чего стимулируется секреция инсулина.
Бигуаниды
увеличивают количество переносчиков глюкозы ГЛЮТ-4 на поверхности мембран клеток жировой ткани и мышц.
Инсулинотерапия
обязательна для СД I типа (1-4 инъекции в день), при СД II типа инсулин иногда назначают для лучшего контроля СД, а также при развитии через 10-15 лет вторичной абсолютной инсулиновой недостаточности.
К перспективным методам лечения сахарного диабета относят следующие: трансплантация островков поджелудочной железы или изолированных β-клеток, трансплантация генетически реконструированных клеток, а также стимуляция регенерации панкреатических островков.
При сахарном диабете обоих типов важнейшее значение имеет диетотерапия. Рекомендуют хорошо сбалансированную диету: на долю углеводов должно приходиться 50—60% общей калорийности пиши (исключение должны составлять легкоусвояемые углеводы, пиво, спиртные напитки, сиропы, пирожные и др.); на долю белков — 15—20%; на долю всех жиров — не более 25-30%. Пищу следует принимать 5—6 раз в течение суток.
Литература:
И.И. Дедов., Г.А. Мельниченко, В.В. Фадеев. Эндокринология. Москва.: «Медицина». 2000г.
|